当前位置:
X-MOL 学术
›
J. Mol. Recognit.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Systematic inhibitor selectivity between PARP1 and PARP2 enzymes: Molecular implications for ovarian cancer personalized therapy
Journal of Molecular Recognition ( IF 2.7 ) Pub Date : 2021-03-08 , DOI: 10.1002/jmr.2891 Xueqian Zuo 1 , Haibo Zhao 1 , Dan Li 1
Journal of Molecular Recognition ( IF 2.7 ) Pub Date : 2021-03-08 , DOI: 10.1002/jmr.2891 Xueqian Zuo 1 , Haibo Zhao 1 , Dan Li 1
Affiliation
|
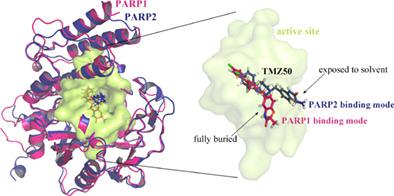
Human poly(ADP-ribose) polymerases (PARPs) are a class of nuclear enzymes involved in the pathogenesis of diverse gynecologic tumors. The PARP1 and PARP2 are the two most documented members in PARP family, which have been approved as the druggable targets of ovarian and cervical cancers. Selective targeting of the two enzymes with small-molecule inhibitors is a great challenge due to the high conservation in catalytic domain and active site. Here, we investigate the systematic selectivity profile of sophisticated PARP inhibitors between the two enzymes. Computational methods are used to model/optimize the complex structures of inhibitor ligands with PARP1/2 catalytic domains and then to estimate the theoretical Fenzymatic assays exhibit a good consistence with theoretical selectivity over six tested inhibitor samples (rc2 = 0.857). It is revealed that the inhibitor selectivity is conferred from the exquisite difference in the residue composition and structural architecture of both the local activity sites and the whole catalytic domains of the two enzymes. In particular, the TMZ50 and ME0328 show strong selectivity between PARP1 and PARP2, but only the former has a potent activity on the two enzymes, whereas the latter can only inhibit the enzymes moderately. These compounds can be considered as potential lead molecular entities to develop new specific PARP-selective inhibitor drugs for personalized therapy combating gynecologic cancers.
中文翻译:

PARP1和PARP2酶之间的系统抑制剂选择性:卵巢癌个体化治疗的分子意义
人聚(ADP-核糖)聚合酶(PARPs)是一类参与多种妇科肿瘤发病机制的核酶。PARP1 和 PARP2 是 PARP 家族中记录最多的两个成员,已被批准为卵巢癌和宫颈癌的药物靶点。由于催化结构域和活性位点的高度保守性,用小分子抑制剂选择性靶向这两种酶是一个巨大的挑战。在这里,我们研究了两种酶之间复杂的 PARP 抑制剂的系统选择性特征。计算方法用于模拟/优化具有 PARP1/2 催化结构域的抑制剂配体的复杂结构,然后估计理论上的 Fenzymatic 测定在六个测试的抑制剂样品中表现出与理论选择性良好的一致性(rc 2 = 0.857)。结果表明,抑制剂的选择性是由两种酶的局部活性位点和整个催化结构域的残基组成和结构结构的细微差异赋予的。特别是TMZ50和ME0328在PARP1和PARP2之间表现出很强的选择性,但只有前者对这两种酶有很强的活性,而后者只能对这两种酶有适度的抑制作用。这些化合物可被视为潜在的先导分子实体,以开发新的特异性 PARP 选择性抑制剂药物,用于针对妇科癌症的个体化治疗。
更新日期:2021-03-08
中文翻译:
PARP1和PARP2酶之间的系统抑制剂选择性:卵巢癌个体化治疗的分子意义
人聚(ADP-核糖)聚合酶(PARPs)是一类参与多种妇科肿瘤发病机制的核酶。PARP1 和 PARP2 是 PARP 家族中记录最多的两个成员,已被批准为卵巢癌和宫颈癌的药物靶点。由于催化结构域和活性位点的高度保守性,用小分子抑制剂选择性靶向这两种酶是一个巨大的挑战。在这里,我们研究了两种酶之间复杂的 PARP 抑制剂的系统选择性特征。计算方法用于模拟/优化具有 PARP1/2 催化结构域的抑制剂配体的复杂结构,然后估计理论上的 Fenzymatic 测定在六个测试的抑制剂样品中表现出与理论选择性良好的一致性(rc 2 = 0.857)。结果表明,抑制剂的选择性是由两种酶的局部活性位点和整个催化结构域的残基组成和结构结构的细微差异赋予的。特别是TMZ50和ME0328在PARP1和PARP2之间表现出很强的选择性,但只有前者对这两种酶有很强的活性,而后者只能对这两种酶有适度的抑制作用。这些化合物可被视为潜在的先导分子实体,以开发新的特异性 PARP 选择性抑制剂药物,用于针对妇科癌症的个体化治疗。


























 京公网安备 11010802027423号
京公网安备 11010802027423号