当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Conformational Strain Indicated by Ramachandran Angles for the Protein Backbone Is Only Weakly Related to the Flexibility
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2021-03-05 , DOI: 10.1021/acs.jpcb.1c00168 Ashraya Ravikumar 1 , Alexandre G. de Brevern 2, 3, 4, 5 , Narayanaswamy Srinivasan 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2021-03-05 , DOI: 10.1021/acs.jpcb.1c00168 Ashraya Ravikumar 1 , Alexandre G. de Brevern 2, 3, 4, 5 , Narayanaswamy Srinivasan 1
Affiliation
|
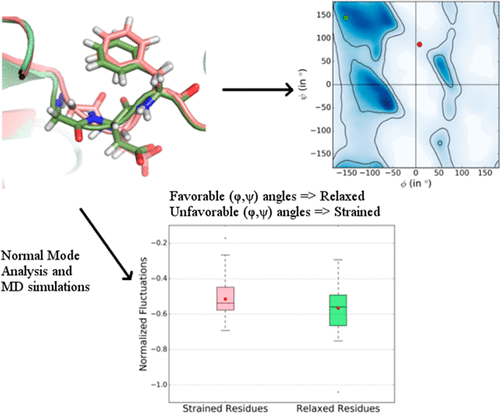
Studies on energy associated with free dipeptides have shown that conformers with unfavorable (ϕ,ψ) torsion angles have higher energy compared to conformers with favorable (ϕ,ψ) angles. It is expected that higher energy confers higher dynamics and flexibility to that part of the protein. Here, we explore a potential relationship between conformational strain in a residue due to unfavorable (ϕ,ψ) angles and its flexibility and dynamics in the context of protein structures. We compared flexibility of strained and relaxed residues, which are recognized based on outlier/allowed and favorable (ϕ,ψ) angles respectively, using normal-mode analysis (NMA). We also performed in-depth analysis on flexibility and dynamics at catalytic residues in protein kinases, which exhibit different strain status in different kinase structures using NMA and molecular dynamics simulations. We underline that strain of a residue, as defined by backbone torsion angles, is almost unrelated to the flexibility and dynamics associated with it. Even the overall trend observed among all high-resolution structures in which relaxed residues tend to have slightly higher flexibility than strained residues is counterintuitive. Consequently, we propose that identifying strained residues based on (ϕ,ψ) values is not an effective way to recognize energetic strain in protein structures.
中文翻译:

Ramachandran角指示蛋白质骨架的构象应变仅与柔韧性相关
对与自由二肽相关的能量的研究表明,与具有有利(ϕ,ψ)角的构象异构体相比,具有不利(ϕ,ψ)扭转角的构象异构体具有更高的能量。期望更高的能量赋予该蛋白质的该部分更高的动力学和灵活性。在这里,我们探讨了由于不利的(ϕ,ψ)角而在残基中的构象应变与其在蛋白质结构中的柔韧性和动力学之间的潜在关系。我们使用正常模式分析(NMA)比较了应变残基和松弛残基的柔韧性,分别根据离群值/允许残差和有利(φ,ψ)角度进行识别。我们还对蛋白激酶中催化残基的灵活性和动力学进行了深入分析,使用NMA和分子动力学模拟,它们在不同的激酶结构中表现出不同的应变状态。我们强调,由骨架扭转角定义的残基应变几乎与其相关的灵活性和动态性无关。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。
更新日期:2021-03-18
中文翻译:
Ramachandran角指示蛋白质骨架的构象应变仅与柔韧性相关
对与自由二肽相关的能量的研究表明,与具有有利(ϕ,ψ)角的构象异构体相比,具有不利(ϕ,ψ)扭转角的构象异构体具有更高的能量。期望更高的能量赋予该蛋白质的该部分更高的动力学和灵活性。在这里,我们探讨了由于不利的(ϕ,ψ)角而在残基中的构象应变与其在蛋白质结构中的柔韧性和动力学之间的潜在关系。我们使用正常模式分析(NMA)比较了应变残基和松弛残基的柔韧性,分别根据离群值/允许残差和有利(φ,ψ)角度进行识别。我们还对蛋白激酶中催化残基的灵活性和动力学进行了深入分析,使用NMA和分子动力学模拟,它们在不同的激酶结构中表现出不同的应变状态。我们强调,由骨架扭转角定义的残基应变几乎与其相关的灵活性和动态性无关。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。即使在所有高分辨率结构中观察到的总体趋势(其中松弛的残基往往比应变的残基具有略高的柔韧性)也是违反直觉的。因此,我们建议基于(ϕ,ψ)值识别应变残基不是识别蛋白质结构中高能应变的有效方法。

























 京公网安备 11010802027423号
京公网安备 11010802027423号