当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Advancing Rational Control of Peptide–Surface Complexes
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2021-03-04 , DOI: 10.1021/acs.jpcb.0c10740 Siva Dasetty 1 , Sapna Sarupria 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2021-03-04 , DOI: 10.1021/acs.jpcb.0c10740 Siva Dasetty 1 , Sapna Sarupria 1
Affiliation
|
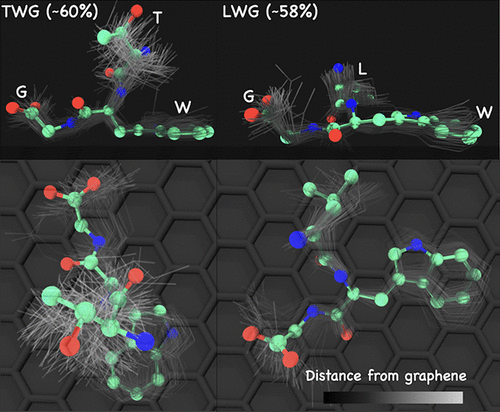
Understanding peptide–surface interactions is crucial for programming self-assembly of peptides at surfaces and in realizing their applications, such as biosensors and biomimetic materials. In this study, we developed insights into the dependence of a residue’s interaction with a surface on its neighboring residue in a tripeptide using molecular dynamics simulations. This knowledge is integral for designing rational mutations to control peptide–surface complexes. Using graphene as our model surface, we estimated the free energy of adsorption (ΔAads) and extracted predominant conformations of 26 tripeptides with the motif LNR–CR–Gly, where LNR and CR are variable left-neighboring and central residues, respectively. We considered a combination of strongly adsorbing (Phe, Trp, and Arg) and weakly adsorbing (Ala, Val, Leu, Ser, and Thr) amino acids on graphene identified in a prior study to form the tripeptides. Our results indicate that ΔAads of a tripeptide cannot be estimated as the sum of ΔAads of each residue indicating that the residues in a tripeptide do not behave as independent entities. We observed that the contributions from the strongly adsorbing amino acids were dominant, which suggests that such residues could be used for strengthening peptide–graphene interactions irrespective of their neighboring residues. In contrast, the adsorption of weakly adsorbing central residues is dependent on their neighboring residues. Our structural analysis revealed that the dihedral angles of LNR are more correlated with that of CR in the adsorbed state than in bulk state. Together with ΔAads trends, this implies that different backbone structures of a given CR can be accessed for a similar ΔAads by varying the LNR. Therefore, incorporation of context effects in designing mutations can lead to desired peptide structure at surfaces. Our results also emphasize that these cooperative effects in ΔAads and structure are not easily predicted a priori. The collective results have applications in guiding rational mutagenesis techniques to control orientation of peptides at surfaces and in developing peptide structure prediction algorithms in adsorbed state from its sequence.
中文翻译:

推进对肽-表面复合物的合理控制
了解肽与表面的相互作用对于编程肽在表面的自组装以及实现其应用(例如生物传感器和仿生材料)至关重要。在这项研究中,我们使用分子动力学模拟对三肽中残基与表面的相互作用及其相邻残基的依赖性进行了深入研究。这些知识是设计合理突变以控制肽表面复合物所不可或缺的。使用石墨烯作为我们的模型表面,我们估计吸附(Δ的自由能一广告)并提取了26个三肽的主要构象,其基序为LNR–CR–Gly,其中LNR和CR分别为可变的左邻和中心残基。我们考虑了在先前研究中鉴定出的形成三肽的强吸附(Phe,Trp和Arg)和弱吸附(Ala,Val,Leu,Ser和Thr)氨基酸的组合。我们的研究结果表明,Δ一个广告三肽的无法估计为Δ的总和一个广告每个残基的“残基”表示三肽中的残基不充当独立实体。我们观察到强吸附氨基酸的贡献占主导地位,这表明此类残基可用于增强肽-石墨烯相互作用,而与它们的相邻残基无关。相反,弱吸附的中心残基的吸附取决于它们相邻的残基。我们的结构分析表明,在吸附状态下,LNR的二面角与CR的二面角比在本体状态下的二面角更相关。加上Δ一个广告的发展趋势,这意味着一个给定的CR的,不同的骨架结构可以为类似Δ访问一个广告通过改变LNR。因此,在设计突变中引入上下文效应可以导致表面上所需的肽结构。我们的研究结果还强调,在Δ这些协同效应一个广告和结构不容易预测的先验。总体结果可用于指导合理的诱变技术以控制肽在表面上的方向,并用于开发从其序列处于吸附状态的肽结构预测算法。
更新日期:2021-03-18
中文翻译:
推进对肽-表面复合物的合理控制
了解肽与表面的相互作用对于编程肽在表面的自组装以及实现其应用(例如生物传感器和仿生材料)至关重要。在这项研究中,我们使用分子动力学模拟对三肽中残基与表面的相互作用及其相邻残基的依赖性进行了深入研究。这些知识是设计合理突变以控制肽表面复合物所不可或缺的。使用石墨烯作为我们的模型表面,我们估计吸附(Δ的自由能一广告)并提取了26个三肽的主要构象,其基序为LNR–CR–Gly,其中LNR和CR分别为可变的左邻和中心残基。我们考虑了在先前研究中鉴定出的形成三肽的强吸附(Phe,Trp和Arg)和弱吸附(Ala,Val,Leu,Ser和Thr)氨基酸的组合。我们的研究结果表明,Δ一个广告三肽的无法估计为Δ的总和一个广告每个残基的“残基”表示三肽中的残基不充当独立实体。我们观察到强吸附氨基酸的贡献占主导地位,这表明此类残基可用于增强肽-石墨烯相互作用,而与它们的相邻残基无关。相反,弱吸附的中心残基的吸附取决于它们相邻的残基。我们的结构分析表明,在吸附状态下,LNR的二面角与CR的二面角比在本体状态下的二面角更相关。加上Δ一个广告的发展趋势,这意味着一个给定的CR的,不同的骨架结构可以为类似Δ访问一个广告通过改变LNR。因此,在设计突变中引入上下文效应可以导致表面上所需的肽结构。我们的研究结果还强调,在Δ这些协同效应一个广告和结构不容易预测的先验。总体结果可用于指导合理的诱变技术以控制肽在表面上的方向,并用于开发从其序列处于吸附状态的肽结构预测算法。


























 京公网安备 11010802027423号
京公网安备 11010802027423号