当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Graphical Transition Moment Decomposition and Conceptual Density Functional Theory Approaches to Study the Fundamental and Lower-Level Overtone Absorption Intensities of Some OH Stretching Vibrations
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2021-03-05 , DOI: 10.1021/acs.jpca.0c11619 Masafumi Tsuyuki 1 , Shunki Furudate 1 , Yuto Kugaya 1 , Satoshi Yabushita 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2021-03-05 , DOI: 10.1021/acs.jpca.0c11619 Masafumi Tsuyuki 1 , Shunki Furudate 1 , Yuto Kugaya 1 , Satoshi Yabushita 1
Affiliation
|
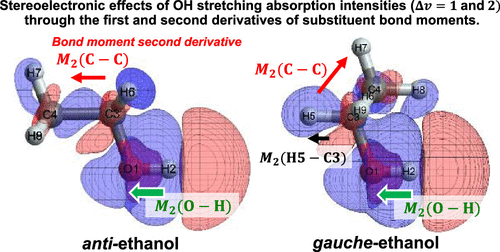
The investigation of electron density migrations caused by molecular structure changes is of central importance in various fields of chemistry. To address this topic in general and to study absorption intensities of vibrations, we analyze sensitive dipole moment functions (DMFs) of a molecule by combining the linear response function of conceptual DFT and bond dipoles separated by the quantum theory of atoms in molecules with a graphical transition moment decomposition scheme. The fundamental intensities of OH stretching vibrations depend strongly on the substituents but only weakly on the molecular conformations. Interestingly, in some alcohols, completely opposite trends have been observed for the lower-level overtone intensities: a weak substituent dependence but a stronger conformation dependence. It is well known that the formation of a hydrogen-bonded complex increases the OH stretching fundamental intensity, but less well known is the decrease in their overtone intensities. To investigate these characteristics comprehensively, we calculated their intensities (Δv = 1, 2, and 3) for conformers of ethanol and trifluoroethanol (TFE) and hydrogen-bonded phenol (PhOH) systems via the DFT method in the local mode model for the OH stretching coordinate ΔR. Their first and second derivatives of the electron density with respect to ΔR were calculated and interpreted using their bond moments. For ethanol and TFE, the OH, CC, and CH bond moments were found to make an important contribution to the molecular DMF derivatives parallel to the OH bond. The OH bond contributes only to the first derivative of DMF, and its conformational dependence is determined by the magnitude of the charge polarization of each structure. The electron density derivatives in the CC bond region were largely maintained during the internal rotation; thus, their conformation-dependent contributions were expressed by a geometrical factor of the CC bond direction. The CH bond at the antiperiplanar position of the OH bond was found to make a remarkably large contribution to the second derivative of DMF in the gauche conformer. The importance of electron density migration on substituents was also identified in the hydrogen-bonded phenol, in which the π-electron density change on the aromatic ring was clearly shown. This migration creates the DMF derivatives both perpendicular and parallel to the OH bond and strongly affects the absorption intensities. In all the cases, some bond moments on the substituents contribute to the first and second DMF derivatives in a structure-dependent manner, thus explaining their stereoelectronic effects.
中文翻译:

图形化过渡矩分解和概念密度泛函理论方法研究某些OH拉伸振动的基本和较低水平的泛音吸收强度
由分子结构变化引起的电子密度迁移的研究在化学的各个领域中至关重要。为了总体上解决这个问题并研究振动的吸收强度,我们通过将概念性DFT的线性响应函数和分子中的原子量子理论所分离的键合偶极子的线性响应函数与图形结合起来,来分析分子的敏感偶极矩函数(DMF)。过渡力矩分解方案。OH拉伸振动的基本强度在很大程度上取决于取代基,而在分子构象方面则微弱。有趣的是,在某些醇中,较低的泛音强度已观察到完全相反的趋势:较弱的取代基依赖性但较强的构象依赖性。众所周知,氢键配合物的形成增加了OH拉伸的基本强度,但鲜为人知的是其泛音强度的降低。为了全面研究这些特征,我们计算了它们的强度(Δv = 1、2和3),用于乙醇和三氟乙醇(TFE)和氢键合苯酚(PhOH)系统的构象异构体,通过DFT方法在局部模式模型中确定OH拉伸坐标ΔR。电子密度相对于ΔR的一阶和二阶导数用它们的键矩来计算和解释。对于乙醇和TFE,发现OH,CC和CH键矩对平行于OH键的分子DMF衍生物起重要作用。OH键仅对DMF的一阶导数起作用,其构象依赖性由每个结构的电荷极化强度决定。在内旋过程中,CC键区的电子密度导数得到了很大的维持;因此,它们的构象依赖性贡献由CC键方向的几何因子表示。发现在OH键的反平面位置上的CH键对gauche构象异构体中的DMF的二阶衍生物具有显着的贡献。在取代氢的苯酚中,还确定了电子密度迁移对取代基的重要性,其中清楚地显示了芳环上的π电子密度变化。这种迁移产生了垂直和平行于OH键的DMF衍生物,并强烈影响吸收强度。在所有情况下,取代基上的某些键矩均以与结构有关的方式有助于第一和第二DMF衍生物,从而解释了它们的立体电子效应。
更新日期:2021-03-18
中文翻译:
图形化过渡矩分解和概念密度泛函理论方法研究某些OH拉伸振动的基本和较低水平的泛音吸收强度
由分子结构变化引起的电子密度迁移的研究在化学的各个领域中至关重要。为了总体上解决这个问题并研究振动的吸收强度,我们通过将概念性DFT的线性响应函数和分子中的原子量子理论所分离的键合偶极子的线性响应函数与图形结合起来,来分析分子的敏感偶极矩函数(DMF)。过渡力矩分解方案。OH拉伸振动的基本强度在很大程度上取决于取代基,而在分子构象方面则微弱。有趣的是,在某些醇中,较低的泛音强度已观察到完全相反的趋势:较弱的取代基依赖性但较强的构象依赖性。众所周知,氢键配合物的形成增加了OH拉伸的基本强度,但鲜为人知的是其泛音强度的降低。为了全面研究这些特征,我们计算了它们的强度(Δv = 1、2和3),用于乙醇和三氟乙醇(TFE)和氢键合苯酚(PhOH)系统的构象异构体,通过DFT方法在局部模式模型中确定OH拉伸坐标ΔR。电子密度相对于ΔR的一阶和二阶导数用它们的键矩来计算和解释。对于乙醇和TFE,发现OH,CC和CH键矩对平行于OH键的分子DMF衍生物起重要作用。OH键仅对DMF的一阶导数起作用,其构象依赖性由每个结构的电荷极化强度决定。在内旋过程中,CC键区的电子密度导数得到了很大的维持;因此,它们的构象依赖性贡献由CC键方向的几何因子表示。发现在OH键的反平面位置上的CH键对gauche构象异构体中的DMF的二阶衍生物具有显着的贡献。在取代氢的苯酚中,还确定了电子密度迁移对取代基的重要性,其中清楚地显示了芳环上的π电子密度变化。这种迁移产生了垂直和平行于OH键的DMF衍生物,并强烈影响吸收强度。在所有情况下,取代基上的某些键矩均以与结构有关的方式有助于第一和第二DMF衍生物,从而解释了它们的立体电子效应。


























 京公网安备 11010802027423号
京公网安备 11010802027423号