Journal of Molecular Graphics and Modelling ( IF 2.9 ) Pub Date : 2021-02-22 , DOI: 10.1016/j.jmgm.2021.107869 A M S Santos 1 , E Moreira 2 , A Meiyazhagan 3 , D L Azevedo 1
|
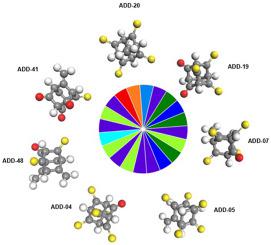
The objective of this work, is to study adamantanes and to tune their bandgap, since pure adamantane is considered as an insulator due to its high bandgap energy. For this, we doped adamantane with oxygen and sulfur atoms, thus obtaining 730 different structures with double bonds and 730 different structures with single bonds, for a total of 1460 structures, and compared their properties. Among all, 31 molecules were selected that best represented the reduced bandgap behavior. The calculations with greater precision in its results were made using the Local Density Approximation (LDA), in the Density-Functional Theory (DFT) formalism, with PWC functional and TNP basis set. The electronic and optical properties were analyzed, by calculating the energy gap and absorption spectrum. Importantly, we observed that molecules doped with sulfur atoms (double bonds) had their energy gap reduced significantly compared to molecules doped with sulfur and/or oxygen atom with single bonds and pristine adamantane. It was found that in the absorption spectrum, the sulfur-doped structures had their spectrum shifted to the visible region, a fact that becomes relevant for potential dyes and optoelectronic applications. From the seven selected functionalized adamantanes (ADD-04, ADD-05, ADD-07, ADD-19, ADD-20, ADD-41, and ADD-48), any of these could be used as a dye. However, the ADD-20 molecule in particular, which presented optical absorption near (RGB) primary colors, could indicate a potential quantum dot material for application in developing screens of various electronic devices.
中文翻译:
键序对氧/硫官能化金刚烷的光电性能的影响
这项工作的目的是研究金刚烷并调整其带隙,因为纯金刚烷由于其高带隙能量而被认为是绝缘体。为此,我们用氧和硫原子掺杂了金刚烷,从而获得了730个具有双键的不同结构和730个具有单键的不同结构,共计1460个结构,并比较了它们的性能。其中,选择了31个最能代表带隙行为减少的分子。使用密度函数理论(DFT)形式主义中的局部密度近似(LDA),并使用PWC函数和TNP基集,可以更精确地计算结果。通过计算能隙和吸收光谱来分析电子和光学性质。重要的,我们观察到,与掺杂有单键和原始金刚烷的硫和/或氧原子的分子相比,掺杂有硫原子(双键)的分子的能隙显着减小。发现在吸收光谱中,硫掺杂结构的光谱移至可见光区域,这一事实与潜在的染料和光电应用有关。从七个选定的功能化金刚烷类化合物(ADD-04,ADD-05,ADD-07,ADD-19,ADD-20,ADD-41和ADD-48)中,任何一种都可用作染料。但是,特别是ADD-20分子,它在(RGB)原色附近表现出光吸收,可能表示一种潜在的量子点材料,可用于开发各种电子设备的屏幕。

























 京公网安备 11010802027423号
京公网安备 11010802027423号