当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Polarization Energies from Efficient Representation of the Linear Density–Density Response Function
Advanced Theory and Simulations ( IF 3.3 ) Pub Date : 2021-01-20 , DOI: 10.1002/adts.202000260 Christian Dreßler 1 , Daniel Sebastiani 1
Advanced Theory and Simulations ( IF 3.3 ) Pub Date : 2021-01-20 , DOI: 10.1002/adts.202000260 Christian Dreßler 1 , Daniel Sebastiani 1
Affiliation
|
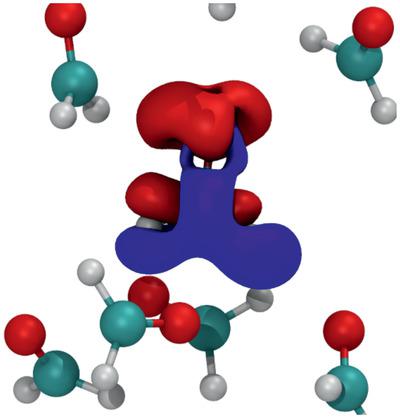
The authors present a proof‐of‐concept study for the calculation of atomic forces on a solvated molecule by means of the linear density–density response function in its moment expanded representation. The density–density response function represents an efficient way to compute molecular forces for arbitrary external potentials via an ab initio scheme, without the need to perform an explicit self‐consistent quantum chemical calculation for each configuration of the chemical environment. Here, the authors show that it is indeed possible to determine the atomic forces of interacting bulk‐like molecular complexes due to polarization effects of the surrounding molecules with good accuracy. This study represents a significant step the practical applicability of the approach, which is still in a development phase. The potential application of the computational scheme in terms of molecular dynamics simulations is illustrated by considering a variety of cluster conformations, as they would be found within a molecular dynamics trajectory.
中文翻译:

线性密度-密度响应函数的有效表示的极化能量
作者提出了一个概念证明研究,用于通过其瞬间扩展表示中的线性密度-密度响应函数来计算溶剂化分子上的原子力。密度-密度响应函数代表了一种通过从头算方案计算任意外部电位的分子力的有效方法,而无需为每种化学环境进行明确的自洽量子化学计算。在这里,作者表明,由于周围分子的极化效应,确实有可能确定相互作用的块状分子复合物的原子力。这项研究代表了该方法的实际适用性的重要一步,该方法仍处于开发阶段。
更新日期:2021-01-20
中文翻译:
线性密度-密度响应函数的有效表示的极化能量
作者提出了一个概念证明研究,用于通过其瞬间扩展表示中的线性密度-密度响应函数来计算溶剂化分子上的原子力。密度-密度响应函数代表了一种通过从头算方案计算任意外部电位的分子力的有效方法,而无需为每种化学环境进行明确的自洽量子化学计算。在这里,作者表明,由于周围分子的极化效应,确实有可能确定相互作用的块状分子复合物的原子力。这项研究代表了该方法的实际适用性的重要一步,该方法仍处于开发阶段。


























 京公网安备 11010802027423号
京公网安备 11010802027423号