当前位置:
X-MOL 学术
›
Biopolymers
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A high-throughput predictive method for sequence-similar fold switchers
Biopolymers ( IF 2.9 ) Pub Date : 2021-01-19 , DOI: 10.1002/bip.23416 Allen K Kim 1, 2 , Loren L Looger 3 , Lauren L Porter 1, 2
Biopolymers ( IF 2.9 ) Pub Date : 2021-01-19 , DOI: 10.1002/bip.23416 Allen K Kim 1, 2 , Loren L Looger 3 , Lauren L Porter 1, 2
Affiliation
|
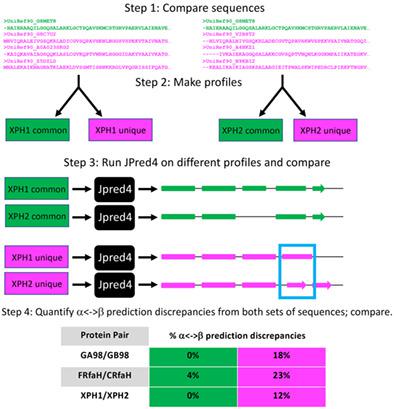
Although most experimentally characterized proteins with similar sequences assume the same folds and perform similar functions, an increasing number of exceptions is emerging. One class of exceptions comprises sequence-similar fold switchers, whose secondary structures shift from α-helix <-> β-sheet through a small number of mutations, a sequence insertion, or a deletion. Predictive methods for identifying sequence-similar fold switchers are desirable because some are associated with disease and/or can perform different functions in cells. Here, we use homology-based secondary structure predictions to identify sequence-similar fold switchers from their amino acid sequences alone. To do this, we predicted the secondary structures of sequence-similar fold switchers using three different homology-based secondary structure predictors: PSIPRED, JPred4, and SPIDER3. We found that α-helix <-> β-strand prediction discrepancies from JPred4 discriminated between the different conformations of sequence-similar fold switchers with high statistical significance (P < 1.8*10−19). Thus, we used these discrepancies as a classifier and found that they can often robustly discriminate between sequence-similar fold switchers and sequence-similar proteins that maintain the same folds (Matthews Correlation Coefficient of 0.82). We found that JPred4 is a more robust predictor of sequence-similar fold switchers because of (a) the curated sequence database it uses to produce multiple sequence alignments and (b) its use of sequence profiles based on Hidden Markov Models. Our results indicate that inconsistencies between JPred4 secondary structure predictions can be used to identify some sequence-similar fold switchers from their sequences alone. Thus, the negative information from inconsistent secondary structure predictions can potentially be leveraged to identify sequence-similar fold switchers from the broad base of genomic sequences.
中文翻译:

序列相似折叠切换器的高通量预测方法
尽管大多数经过实验表征的具有相似序列的蛋白质具有相同的折叠并执行相似的功能,但越来越多的例外情况正在出现。一类例外包括序列相似的折叠转换蛋白,其二级结构通过少量突变、序列插入或缺失从α-螺旋<->β-折叠转变。用于鉴定序列相似折叠转换蛋白的预测方法是可取的,因为有些与疾病相关和/或可以在细胞中执行不同的功能。在这里,我们使用基于同源性的二级结构预测来仅从氨基酸序列中识别序列相似的折叠转换蛋白。为此,我们使用三种不同的基于同源性的二级结构预测因子:PSIPRED、JPred4 和 SPIDER3 来预测序列相似折叠转换蛋白的二级结构。我们发现 JPred4 的 α 螺旋 <-> β 链预测差异区分了序列相似折叠转换子的不同构象,具有高统计显着性 ( P < 1.8*10 -19 )。因此,我们使用这些差异作为分类器,发现它们通常可以稳健地区分序列相似的折叠转换蛋白和保持相同折叠的序列相似的蛋白质(马修斯相关系数为 0.82)。我们发现 JPred4 是序列相似折叠切换器的更稳健的预测器,因为(a)它使用精心策划的序列数据库来产生多个序列比对,以及(b)它使用基于隐马尔可夫模型的序列概况。我们的结果表明,JPred4 二级结构预测之间的不一致可用于仅从序列中识别一些序列相似的折叠转换蛋白。因此,来自不一致二级结构预测的负面信息有可能被用来从广泛的基因组序列中识别序列相似的折叠转换蛋白。
更新日期:2021-01-19
中文翻译:
序列相似折叠切换器的高通量预测方法
尽管大多数经过实验表征的具有相似序列的蛋白质具有相同的折叠并执行相似的功能,但越来越多的例外情况正在出现。一类例外包括序列相似的折叠转换蛋白,其二级结构通过少量突变、序列插入或缺失从α-螺旋<->β-折叠转变。用于鉴定序列相似折叠转换蛋白的预测方法是可取的,因为有些与疾病相关和/或可以在细胞中执行不同的功能。在这里,我们使用基于同源性的二级结构预测来仅从氨基酸序列中识别序列相似的折叠转换蛋白。为此,我们使用三种不同的基于同源性的二级结构预测因子:PSIPRED、JPred4 和 SPIDER3 来预测序列相似折叠转换蛋白的二级结构。我们发现 JPred4 的 α 螺旋 <-> β 链预测差异区分了序列相似折叠转换子的不同构象,具有高统计显着性 ( P < 1.8*10 -19 )。因此,我们使用这些差异作为分类器,发现它们通常可以稳健地区分序列相似的折叠转换蛋白和保持相同折叠的序列相似的蛋白质(马修斯相关系数为 0.82)。我们发现 JPred4 是序列相似折叠切换器的更稳健的预测器,因为(a)它使用精心策划的序列数据库来产生多个序列比对,以及(b)它使用基于隐马尔可夫模型的序列概况。我们的结果表明,JPred4 二级结构预测之间的不一致可用于仅从序列中识别一些序列相似的折叠转换蛋白。因此,来自不一致二级结构预测的负面信息有可能被用来从广泛的基因组序列中识别序列相似的折叠转换蛋白。


























 京公网安备 11010802027423号
京公网安备 11010802027423号