Journal of Proteomics ( IF 3.3 ) Pub Date : 2021-01-13 , DOI: 10.1016/j.jprot.2021.104116 Sangeetha Ramachandran 1 , Tessamma Thomas 1
|
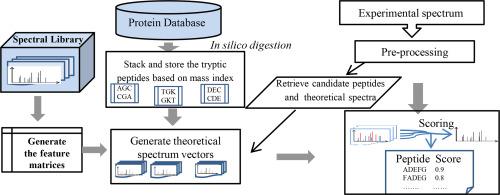
The database search method is a widely accepted method to assign a peptide to the tandem mass spectra. In this study, a new flexible method- FPTMS is introduced to interpret the tandem mass spectra with the known peptide sequences in a protein database. Here the frequency of occurrence of fragment ion peaks extracted from the extensive spectral library is used to predict the theoretical tandem mass spectra of the peptides. The dot product scoring and windowed-xcorr scoring methods were implemented to score the experimental spectrum against the theoretical peptide spectra. Windowed-xcorr is introduced to tackle the mass errors and the cleavage position of the fragmentation process. The new method with windowed-xcorr shows an improved identification rate compared to the existing search engines Crux-Tide and X!Tandem at 1% False Discovery Rate (FDR) for the dataset considered in this study.
Significance
Identifying or sequencing of the peptide from tandem mass spectra is an important application in mass spectrometry-based proteomics. Collision-induced dissociation (CID) fragmentation spectra have been widely used to develop a peptide identification algorithm using database search strategy. CID fragmentation behavior is a complex process and found to have dependency on the sequences of peptide, charge state, and residue content. The inclusion of more features of peptide fragmentation behavior and adaptable scoring algorithm improves the efficiency of the peptide identification algorithm.
中文翻译:
FPTMS:基于频率的方法,可从低能碰撞诱导的解离串联质谱图中鉴定肽
数据库搜索方法是将肽分配给串联质谱的一种广泛接受的方法。在这项研究中,引入了一种新的灵活方法FPTMS来解释蛋白质数据库中已知肽序列的串联质谱。在此,从广泛的质谱库中提取的碎片离子峰的出现频率用于预测肽的理论串联质谱。实施点积计分和窗口xcorr计分方法以对理论肽谱的实验谱进行评分。引入Windowed-xcorr来解决质量误差和碎片化过程的裂解位置。与现有的搜索引擎Crux-Tide和X!相比,使用windowed-xcorr的新方法显示出更高的识别率。
意义
从串联质谱鉴定或测序肽是在基于质谱的蛋白质组学中的重要应用。碰撞诱导解离(CID)碎片光谱已广泛用于开发使用数据库搜索策略的肽段识别算法。CID片段化行为是一个复杂的过程,并且发现它依赖于肽序列,电荷状态和残基含量。包含更多的肽片段化行为特征和适应性评分算法可提高肽段识别算法的效率。

























 京公网安备 11010802027423号
京公网安备 11010802027423号