Journal of Advanced Research ( IF 10.7 ) Pub Date : 2021-01-12 , DOI: 10.1016/j.jare.2021.01.005 Amal Souissi 1 , Mariem Ben Said 1 , Ikhlas Ben Ayed 2, 3 , Ines Elloumi 1 , Amal Bouzid 1 , Mohamed Ali Mosrati 1 , Mehdi Hasnaoui 4 , Malek Belcadhi 1 , Nabil Idriss 4 , Hassen Kamoun 2, 3 , Nourhene Gharbi 2, 3 , Abdullah A Gibriel 5 , Abdelaziz Tlili 6, 7 , Saber Masmoudi 1
|
Introduction
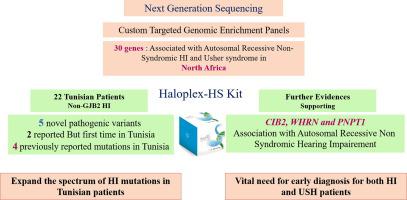
Hearing impairment (HI) is characterized by complex genetic heterogeneity. The evolution of next generation sequencing, including targeted enrichment panels, has revolutionized HI diagnosis.
Objectives
In this study, we investigated genetic causes in 22 individuals with non-GJB2 HI.
Methods
We customized a HaloplexHS kit to include 30 genes known to be associated with autosomal recessive nonsyndromic HI (ARNSHI) and Usher syndrome in North Africa.
Results
In accordance with the ACMG/AMP guidelines, we report 11 pathogenic variants; as follows; five novel variants including three missense (ESRRB-Tyr295Cys, MYO15A-Phe2089Leu and MYO7A-Tyr560Cys) and two nonsense (USH1C-Gln122Ter and CIB2-Arg104Ter) mutations; two previously reported mutations (OTOF-Glu57Ter and PNPT1-Glu475Gly), but first time identified among Tunisian families; and four other identified mutations namely WHRN-Gly808AspfsX11, SLC22A4-Cys113Tyr and two MYO7A compound heterozygous splice site variants that were previously described in Tunisia. Pathogenic variants in WHRN and CIB2 genes, in patients with convincing phenotype ruling out retinitis pigmentosa, provide strong evidence supporting their association with ARNSHI. Moreover, we shed lights on the pathogenic implication of mutations in PNPT1 gene in auditory function providing new evidence for its association with ARNSHI. Lack of segregation of a previously identified causal mutation OTOA-Val603Phe further supports its classification as variant of unknown significance. Our study reports absence of otoacoustic emission in subjects using bilateral hearing aids for several years indicating the importance of screening genetic alteration in OTOF gene for proper management of those patients.
Conclusion
In conclusion, our findings do not only expand the spectrum of HI mutations in Tunisian patients, but also improve our knowledge about clinical relevance of HI causing genes and variants.
中文翻译:
新的致病突变和导致突尼斯人群听力障碍的基因和变异的临床相关性的进一步证据
介绍
听力障碍 (HI) 的特点是复杂的遗传异质性。下一代测序的发展,包括靶向富集面板,彻底改变了 HI 诊断。
目标
在这项研究中,我们调查了 22 名非GJB2 HI个体的遗传原因。
方法
我们定制了 Haloplex HS试剂盒,其中包含 30 个已知与北非常染色体隐性非综合征 HI (ARNSHI) 和 Usher 综合征相关的基因。
结果
根据 ACMG/AMP 指南,我们报告了 11 种致病变异;如下; 五个新变体,包括三个错义(ESRRB- Tyr295Cys、MYO15A -Phe2089Leu 和MYO7A -Tyr560Cys)和两个无义(USH1C- Gln122Ter 和CIB2- Arg104Ter)突变;两个先前报道的突变(OTOF- Glu57Ter 和PNPT1- Glu475Gly),但首次在突尼斯家族中发现;以及其他四个已鉴定的突变,即WHRN- Gly808AspfsX11、SLC22A4 -Cys113Tyr 和两个MYO7A复合杂合剪接位点变体,这些变体先前在突尼斯中有过描述。致病性变异WHRN和CIB2基因在具有排除视网膜色素变性的令人信服的表型的患者中提供了强有力的证据支持它们与 ARNSHI 的关联。此外,我们阐明了PNPT1基因突变在听觉功能中的致病意义,为其与 ARNSHI 的关联提供了新的证据。先前确定的因果突变OTOA -Val603Phe缺乏分离进一步支持其分类为意义未知的变体。我们的研究报告称,多年来使用双侧助听器的受试者没有耳声发射,这表明筛查OTOF基因中的遗传改变对正确管理这些患者的重要性。
结论
总之,我们的研究结果不仅扩大了突尼斯患者的 HI 突变谱,而且提高了我们对 HI 引起基因和变异的临床相关性的了解。


























 京公网安备 11010802027423号
京公网安备 11010802027423号