Catalysis Today ( IF 5.3 ) Pub Date : 2021-01-02 , DOI: 10.1016/j.cattod.2020.12.007 Javier Amaya Suárez , José J. Plata , Antonio M. Márquez , Javier Fdez. Sanz
|
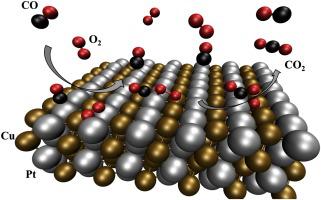
To understand the microscopic mechanism of the CO oxidation reaction at PtCu nanoparticles, which have unique geometric and electronic structures compared to their component metals, we present here a theoretical study, based on density functional theory calculations, of the main reaction steps of this reaction. We examine the O2 dissociation, the CO adsorption and the CO + O2 reaction at an atomic level and use the computed geometries, Bader charges, and vibrational frequencies to rationalize the role of the intermetallic nanoparticles surface structure on the experimentally observed much higher activity of these nanoparticles as catalysts of the preferential oxidation of CO. By comparing with clean Pt (111) surface and with different Cu-doped models of this same surface, our results show that, at the surface, the presence of Cu induces the segregation of CO molecules at Pt sites and of O2 molecules at Cu sites. Contrarily to Pt surfaces, the unassisted O2 dissociation has a high barrier at the intermetallic nanoparticle surface and proceeds through a CO-assisted mechanism in which the new CO bond is formed while the O O bond is broken with a kinetic barrier much lower than on either Pt (111) or in Pt-doped surfaces. The particular structure of the intermetallic surface is shown to have a significant role in the low kinetic barrier for the reaction, allowing for an easy approach of the CO to the adsorbed O2 molecule that permits an early transition state with a low energetic barrier.
O bond is broken with a kinetic barrier much lower than on either Pt (111) or in Pt-doped surfaces. The particular structure of the intermetallic surface is shown to have a significant role in the low kinetic barrier for the reaction, allowing for an easy approach of the CO to the adsorbed O2 molecule that permits an early transition state with a low energetic barrier.
中文翻译:
PtCu金属间化合物对CO氧化的催化活性:理论见解
为了了解 PtCu 纳米粒子 CO 氧化反应的微观机理,与它们的组分金属相比,PtCu 纳米粒子具有独特的几何和电子结构,我们在此基于密度泛函理论计算,对该反应的主要反应步骤进行了理论研究。我们检查了 O 2解离、CO 吸附和 CO + O 2在原子水平上进行反应,并使用计算的几何形状、贝德电荷和振动频率来合理化金属间纳米粒子表面结构对实验观察到的这些纳米粒子作为 CO 优先氧化催化剂的更高活性的作用。 Pt (111) 表面和同一表面的不同 Cu 掺杂模型,我们的结果表明,在表面,Cu 的存在会导致 Pt 位点的 CO 分子和Cu 位点的 O 2分子的分离。与 Pt 表面相反,无辅助的 O 2离解在金属间纳米颗粒表面具有高势垒,并通过 CO 辅助机制进行,其中形成新的 CO 键,而 OO 键断裂的动力学势垒远低于 Pt (111) 或 Pt 掺杂表面。金属间化合物表面的特殊结构显示在反应的低动力势垒中具有重要作用,允许 CO 轻松接近吸附的 O 2分子,从而允许具有低能量势垒的早期过渡态。


























 京公网安备 11010802027423号
京公网安备 11010802027423号