The Protein Journal ( IF 3 ) Pub Date : 2021-01-02 , DOI: 10.1007/s10930-020-09945-6 Mahmoud A A Ibrahim 1 , Alaa H M Abdelrahman 1 , Khaled S Allemailem 2 , Ahmad Almatroudi 2 , Mahmoud F Moustafa 3, 4 , Mohamed-Elamir F Hegazy 5
|
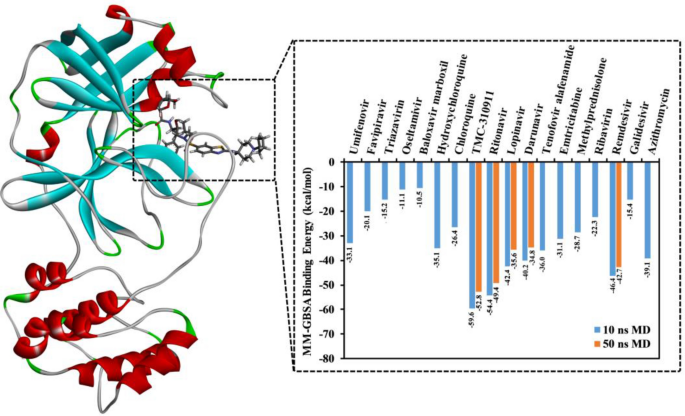
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a recently emanating human infectious coronavirus that causes COVID-19 disease. On 11th March 2020, it has been announced as a pandemic by the World Health Organization (WHO). Recently, several repositioned drugs have been subjected to clinical investigations as anti-COVID-19 drugs. Here, in silico drug discovery tools were utilized to evaluate the binding affinities and features of eighteen anti-COVID-19 drug candidates against SARS-CoV-2 main protease (Mpro). Molecular docking calculations using Autodock Vina showed considerable binding affinities of the investigated drugs with docking scores ranging from − 5.3 to − 8.3 kcal/mol, with higher binding affinities for HIV drugs compared to the other antiviral drugs. Molecular dynamics (MD) simulations were performed for the predicted drug-Mpro complexes for 50 ns, followed by binding energy calculations utilizing molecular mechanics-generalized Born surface area (MM-GBSA) approach. MM-GBSA calculations demonstrated promising binding affinities of TMC-310911 and ritonavir towards SARS-CoV-2 Mpro, with binding energy values of − 52.8 and − 49.4 kcal/mol, respectively. Surpass potentialities of TMC-310911 and ritonavir are returned to their capabilities of forming multiple hydrogen bonds with the proximal amino acids inside Mpro's binding site. Structural and energetic analyses involving root-mean-square deviation, binding energy per-frame, center-of-mass distance, and hydrogen bond length demonstrated the stability of TMC-310911 and ritonavir inside the Mpro's active site over the 50 ns MD simulation. This study sheds light on HIV protease drugs as prospective SARS-CoV-2 Mpro inhibitors.
Graphic Abstract
中文翻译:
对作为潜在 SARS-CoV-2 主要蛋白酶抑制剂的潜在抗 COVID-19 候选药物进行计算机评估
严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 是一种最近出现的人类传染性冠状病毒,可导致 COVID-19 疾病。2020 年 3 月 11 日,它已被世界卫生组织 (WHO) 宣布为大流行病。最近,几种重新定位的药物已作为抗 COVID-19 药物进行了临床研究。在这里,利用计算机药物发现工具评估了 18 种抗 COVID-19 候选药物对 SARS-CoV-2 主要蛋白酶(M pro )的结合亲和力和特征。)。使用 Autodock Vina 进行的分子对接计算显示,与其他抗病毒药物相比,所研究的药物具有相当大的结合亲和力,对接分数范围为 - 5.3 至 - 8.3 kcal/mol,对 HIV 药物的结合亲和力更高。对预测的药物-M pro配合物进行 50 ns 的分子动力学 (MD) 模拟,然后利用分子力学广义玻恩表面积 (MM-GBSA) 方法计算结合能。MM-GBSA 计算表明 TMC-310911 和利托那韦对 SARS-CoV-2 M pro具有良好的结合亲和力,结合能值分别为 - 52.8 和 - 49.4 kcal/mol。TMC-310911 和利托那韦的超越潜力恢复到它们与 M pro结合位点内的近端氨基酸形成多个氢键的能力。涉及均方根偏差、每帧结合能、质心距离和氢键长度的结构和能量分析表明,TMC-310911 和利托那韦在 M pro的活性位点内的稳定性超过 50 ns MD模拟。这项研究揭示了 HIV 蛋白酶药物作为潜在的 SARS-CoV-2 M pro抑制剂的作用。
























 京公网安备 11010802027423号
京公网安备 11010802027423号