Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2020-12-29 , DOI: 10.1016/j.csbj.2020.12.028 Abhinit Kumar 1 , Saurabh Loharch 1 , Sunil Kumar 1 , Rajesh P Ringe 1 , Raman Parkesh 1, 2
|
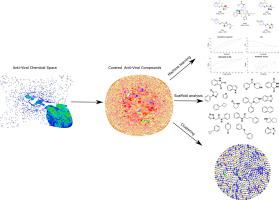
The current life-threatening and tenacious pandemic eruption of coronavirus disease in 2019 (COVID-19) has posed a significant global hazard concerning high mortality rate, economic meltdown, and everyday life distress. The rapid spread of COVID-19 demands countermeasures to combat this deadly virus. Currently, there are no drugs approved by the FDA to treat COVID-19. Therefore, discovering small molecule therapeutics for treating COVID-19 infection is essential. So far, only a few small molecule inhibitors are reported for coronaviruses. There is a need to expand the small chemical space of coronaviruses inhibitors by adding potent and selective scaffolds with anti-COVID activity. In this context, the huge antiviral chemical space already available can be analysed using cheminformatic and machine learning to unearth new scaffolds. We created three specific datasets called “antiviral dataset” (N = 38,428) “drug-like antiviral dataset” (N = 20,963) and “anticorona dataset” (N = 433) for this purpose. We analyzed the 433 molecules of “anticorona dataset” for their scaffold diversity, physicochemical distributions, principal component analysis, activity cliffs, R-group decomposition, and scaffold mapping. The scaffold diversity of the “anticorona dataset” in terms of Murcko scaffold analysis demonstrates a thorough representation of diverse chemical scaffolds. However, physicochemical descriptor analysis and principal component analysis demonstrated negligible drug-like features for the “anticorona dataset” molecules. The “antiviral dataset” and “drug-like antiviral dataset” showed low scaffold diversity as measured by the Gini coefficient. The hierarchical clustering of the “antiviral dataset” against the “anticorona dataset” demonstrated little molecular similarity. We generated a library of frequent fragments and polypharmacological ligands targeting various essential viral proteins such as main protease, helicase, papain-like protease, and replicase polyprotein 1ab. Further structural and chemical features of the “anticorona dataset” were compared with SARS-CoV-2 repurposed drugs, FDA-approved drugs, natural products, and drugs currently in clinical trials. Using machine learning tool DCA (DMax Chemistry Assistant), we converted the “anticorona dataset” into an elegant hypothesis with significant functional biological relevance. Machine learning analysis uncovered that FDA approved drugs, Tizanidine HCl, Cefazolin, Raltegravir, Azilsartan, Acalabrutinib, Luliconazole, Sitagliptin, Meloxicam (Mobic), Succinyl sulfathiazole, Fluconazole, and Pranlukast could be repurposed as effective drugs for COVID-19. Fragment-based scaffold analysis and R-group decomposition uncovered pyrrolidine and the indole molecular scaffolds as the potent fragments for designing and synthesizing the novel drug-like molecules for targeting SARS-CoV-2. This comprehensive and systematic assessment of small-molecule viral therapeutics' entire chemical space realised critical insights to potentially privileged scaffolds that could aid in enrichment and rapid discovery of efficacious antiviral drugs for COVID-19.
中文翻译:
利用化学信息学和机器学习来探索 SARS-CoV-2 潜在小分子抑制剂的可用化学空间
当前,2019 年冠状病毒病 (COVID-19) 威胁生命且顽强地大流行,造成了高死亡率、经济崩溃和日常生活困扰等重大全球危害。COVID-19 的迅速传播需要采取对策来对抗这种致命病毒。目前,FDA 还没有批准治疗 COVID-19 的药物。因此,发现治疗 COVID-19 感染的小分子疗法至关重要。到目前为止,只有少数针对冠状病毒的小分子抑制剂被报道。需要通过添加具有抗新冠活性的有效且选择性的支架来扩大冠状病毒抑制剂的小化学空间。在这种情况下,可以使用化学信息学和机器学习来分析现有的巨大抗病毒化学空间,以发现新的支架。为此,我们创建了三个特定数据集,称为“抗病毒数据集”(N = 38,428)、“类药物抗病毒数据集”(N = 20,963)和“抗新冠数据集”(N = 433)。我们分析了“anticorona 数据集”的 433 个分子的支架多样性、理化分布、主成分分析、活性悬崖、R 基团分解和支架映射。根据 Murcko 支架分析,“抗冠状数据集”的支架多样性展示了多种化学支架的全面表征。然而,物理化学描述符分析和主成分分析表明“抗冠状数据集”分子的药物样特征可以忽略不计。根据基尼系数测量,“抗病毒数据集”和“类药物抗病毒数据集”显示支架多样性较低。“抗病毒数据集”与“抗冠状病毒数据集”的层次聚类显示出很少的分子相似性。我们生成了一个针对各种必需病毒蛋白(例如主蛋白酶、解旋酶、木瓜蛋白酶样蛋白酶和复制酶多蛋白 1ab)的频繁片段和多药理配体文库。将“抗冠状病毒数据集”的进一步结构和化学特征与 SARS-CoV-2 改造药物、 FDA 批准的药物、天然产物和目前处于临床试验中的药物进行了比较。使用机器学习工具 DCA(DMax Chemistry Assistant),我们将“抗冠状数据集”转换为具有重要功能生物学相关性的优雅假设。机器学习分析发现,FDA 批准的药物,盐酸替扎尼定、头孢唑林、拉特拉韦、阿齐沙坦、Acalabrutinib、卢立康唑、西格列汀、美洛昔康 (Mobic)、琥珀酰磺胺噻唑、氟康唑和普兰鲁司特可以重新用作治疗 COVID-19 的有效药物。基于片段的支架分析和 R 基团分解发现吡咯烷和吲哚分子支架是设计和合成针对 SARS-CoV-2 的新型药物样分子的有效片段。

























 京公网安备 11010802027423号
京公网安备 11010802027423号