当前位置:
X-MOL 学术
›
Chem. Res. Toxicol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Impact of Sequencing Depth and Library Preparation on Toxicological Interpretation of RNA-Seq Data in a “Three-Sample” Scenario
Chemical Research in Toxicology ( IF 4.1 ) Pub Date : 2020-12-23 , DOI: 10.1021/acs.chemrestox.0c00368 Dongying Li 1 , Binsheng Gong 1 , Joshua Xu 1 , Baitang Ning 1 , Weida Tong 1
Chemical Research in Toxicology ( IF 4.1 ) Pub Date : 2020-12-23 , DOI: 10.1021/acs.chemrestox.0c00368 Dongying Li 1 , Binsheng Gong 1 , Joshua Xu 1 , Baitang Ning 1 , Weida Tong 1
Affiliation
|
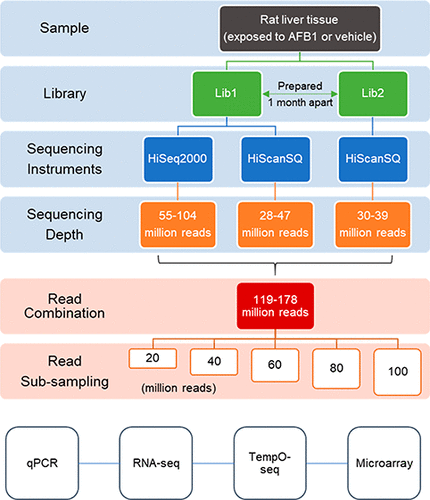
While RNA-sequencing (RNA-seq) has emerged as a standard approach in toxicogenomics, its full potential in gaining underlying toxicological mechanisms is still not clear when only three biological replicates are used. This “three-sample” study design is common in toxicological research, particularly in animal studies during preclinical drug development. Sequencing depth (the total number of reads in an experiment) and library preparation are critical to the resolution and integrity of RNA-seq data and biological interpretation. We used aflatoxin b1 (AFB1), a model toxicant, to investigate the effect of sequencing depth and library preparation in RNA-seq on toxicological interpretation in the “three-sample” scenario. We also compared different gene profiling platforms (RNA-seq, TempO-seq, microarray, and qPCR) using identical liver samples. Well-established mechanisms of AFB1 toxicity served as ground truth for our comparative analyses. We found that a minimum of 20 million reads was sufficient to elicit key toxicity functions and pathways underlying AFB1-induced liver toxicity using three replicates and that identification of differentially expressed genes was positively associated with sequencing depth to a certain extent. Further, our results showed that RNA-seq revealed toxicological insights from pathway enrichment with overall higher statistical power and overlap ratio, compared with TempO-seq and microarray. Moreover, library preparation using the same methods was important to reproducing the toxicological interpretation.
中文翻译:

“三样本”情景中测序深度和文库制备对 RNA-Seq 数据毒理学解释的影响
虽然 RNA 测序 (RNA-seq) 已成为毒物基因组学的标准方法,但当仅使用三个生物学重复时,其在获得潜在毒理学机制方面的全部潜力仍不清楚。这种“三样本”研究设计在毒理学研究中很常见,特别是在临床前药物开发期间的动物研究中。测序深度(实验中的读取总数)和文库制备对于 RNA-seq 数据和生物学解释的分辨率和完整性至关重要。我们使用模型毒物黄曲霉毒素 b1 (AFB1) 来研究 RNA-seq 中测序深度和文库制备对“三样本”情景中毒理学解释的影响。我们还使用相同的肝脏样本比较了不同的基因分析平台(RNA-seq、TempO-seq、微阵列和 qPCR)。公认的 AFB1 毒性机制作为我们比较分析的基本事实。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。
更新日期:2021-02-15
中文翻译:
“三样本”情景中测序深度和文库制备对 RNA-Seq 数据毒理学解释的影响
虽然 RNA 测序 (RNA-seq) 已成为毒物基因组学的标准方法,但当仅使用三个生物学重复时,其在获得潜在毒理学机制方面的全部潜力仍不清楚。这种“三样本”研究设计在毒理学研究中很常见,特别是在临床前药物开发期间的动物研究中。测序深度(实验中的读取总数)和文库制备对于 RNA-seq 数据和生物学解释的分辨率和完整性至关重要。我们使用模型毒物黄曲霉毒素 b1 (AFB1) 来研究 RNA-seq 中测序深度和文库制备对“三样本”情景中毒理学解释的影响。我们还使用相同的肝脏样本比较了不同的基因分析平台(RNA-seq、TempO-seq、微阵列和 qPCR)。公认的 AFB1 毒性机制作为我们比较分析的基本事实。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们发现至少 2000 万个读数足以使用三个重复引发 AFB1 诱导的肝毒性的关键毒性功能和途径,并且差异表达基因的鉴定在一定程度上与测序深度呈正相关。此外,我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。我们的结果表明,与 TempO-seq 和微阵列相比,RNA-seq 揭示了通路富集的毒理学见解,具有更高的统计能力和重叠率。此外,使用相同方法制备文库对于重现毒理学解释很重要。

























 京公网安备 11010802027423号
京公网安备 11010802027423号