当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Enhanced Sampling Path Integral Methods Using Neural Network Potential Energy Surfaces with Application to Diffusion in Hydrogen Hydrates
Advanced Theory and Simulations ( IF 3.3 ) Pub Date : 2020-12-18 , DOI: 10.1002/adts.202000258 Joseph R. Cendagorta 1 , Hengyuan Shen 2 , Zlatko Bačić 1, 3 , Mark E. Tuckerman 1, 3, 4
Advanced Theory and Simulations ( IF 3.3 ) Pub Date : 2020-12-18 , DOI: 10.1002/adts.202000258 Joseph R. Cendagorta 1 , Hengyuan Shen 2 , Zlatko Bačić 1, 3 , Mark E. Tuckerman 1, 3, 4
Affiliation
|
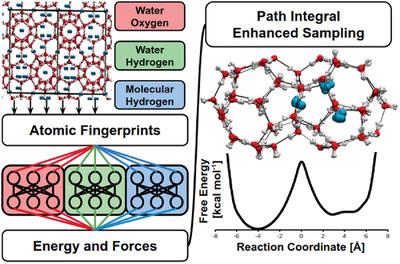
The high cost of ab initio molecular dynamics (AIMD) simulations to model complex physical and chemical systems limits its ability to address many key questions. However, new machine learning‐based representations of complex potential energy surfaces have been introduced in recent years to circumvent computationally demanding AIMD simulations while retaining the same level of accuracy. As these machine learning methods gain in popularity over the next decade, it is important to address the appropriate way to develop and integrate them with well‐established simulation methods. This paper details the parameterization and training, using accurate electronic structure calculations, of artificial neural network potentials (NNPs) to model the intermolecular interactions in a hydrogen clathrate hydrate, wherein hydrogen molecules are confined inside the cavities of the 3D crystalline framework of hydrogen‐bonded water molecules. This new NNP is used in conjunction with new path‐integral based enhanced sampling methods, for inclusion of nuclear quantum effects and promotion of free energy barrier crossing, in order to determine properties that are important for understanding the diffusion of hydrogen gas in the clathrate system. These simulations demonstrate the influence of cage occupancy on the free energy barriers that determine the diffusivity of hydrogen gas through the network of large cages.
中文翻译:

使用神经网络势能面的增强采样路径积分方法及其在氢水合物扩散中的应用
从头开始分子动力学(AIMD)仿真来建模复杂的物理和化学系统的高昂成本限制了它解决许多关键问题的能力。但是,近年来引入了新的基于机器学习的复杂势能面表示方法,以在保持相同精度水平的情况下规避对计算要求苛刻的AIMD仿真。随着这些机器学习方法在未来十年中变得越来越流行,重要的是找到适当的方法来开发它们并将它们与成熟的仿真方法集成在一起。本文详细介绍了使用精确的电子结构计算对人工神经网络电势(NNP)进行参数化和训练,以模拟包合物氢水合物中的分子间相互作用的过程,其中氢分子被限制在氢键水分子的3D晶体框架的腔内。此新NNP与基于路径积分的新增强采样方法结合使用,以包含核量子效应和促进自由能垒的穿越,从而确定对于了解氢在包合物系统中的扩散至关重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。包括核量子效应和促进自由能垒的穿越,以便确定对于了解氢在包合物系统中的扩散非常重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。包括核量子效应和促进自由能垒的穿越,以便确定对于了解氢在包合物系统中的扩散非常重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。
更新日期:2020-12-18
中文翻译:
使用神经网络势能面的增强采样路径积分方法及其在氢水合物扩散中的应用
从头开始分子动力学(AIMD)仿真来建模复杂的物理和化学系统的高昂成本限制了它解决许多关键问题的能力。但是,近年来引入了新的基于机器学习的复杂势能面表示方法,以在保持相同精度水平的情况下规避对计算要求苛刻的AIMD仿真。随着这些机器学习方法在未来十年中变得越来越流行,重要的是找到适当的方法来开发它们并将它们与成熟的仿真方法集成在一起。本文详细介绍了使用精确的电子结构计算对人工神经网络电势(NNP)进行参数化和训练,以模拟包合物氢水合物中的分子间相互作用的过程,其中氢分子被限制在氢键水分子的3D晶体框架的腔内。此新NNP与基于路径积分的新增强采样方法结合使用,以包含核量子效应和促进自由能垒的穿越,从而确定对于了解氢在包合物系统中的扩散至关重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。包括核量子效应和促进自由能垒的穿越,以便确定对于了解氢在包合物系统中的扩散非常重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。包括核量子效应和促进自由能垒的穿越,以便确定对于了解氢在包合物系统中的扩散非常重要的性质。这些模拟证明了笼子占用对自由能垒的影响,自由能垒决定了氢气通过大笼子网络的扩散率。

























 京公网安备 11010802027423号
京公网安备 11010802027423号