当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Efficient Calculation of Small Molecule Binding in Metal–Organic Frameworks and Porous Organic Cages
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jpcc.0c08617 Sebastian Spicher 1 , Markus Bursch 1 , Stefan Grimme 1
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-12-03 , DOI: 10.1021/acs.jpcc.0c08617 Sebastian Spicher 1 , Markus Bursch 1 , Stefan Grimme 1
Affiliation
|
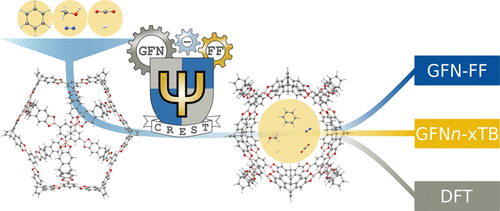
The activation, storage, and separation of gases and fuels are closely related to the reduction of greenhouse gas emissions, the widespread use of renewable energies, and the application of industrial gases. Metal–organic frameworks (MOF) and porous organic cages (POC) are an emerging class of crystalline porous materials that show promising characteristics in this field. Yet, their accurate theoretical description poses a challenge to existing methods due to the sheer size of the pores and cages as well as their often complex structure. In this work, the performance of generally applicable density functional approximations (DFAs), semiempirical quantum mechanical (SQM) methods, and force fields (FFs) for the calculation of binding energies of various gases in molecular cutouts of MOFs and POCs is tested with reference to high-level PBE0-D4/def2-TZVP hybrid DFT energies. Therefore, favorable binding sites for greenhouse gases (CO2), energy-related gases (H2, methanol, and benzene), and industrial gases (N2) are determined by an efficient conformer search algorithm (CREST). The resulting structures are further optimized by DFT (B97-3c), semiempirical (GFN2-xTB), and force-field (GFN-FF) methods to yield the binding sites and corresponding energies. With mean absolute deviations ranging from 1.1 to 1.4 kcal mol–1 for all tested systems, the considered GFN methods reach an accuracy remarkably close to the DFT reference, justifying their application for efficient binding site screening. In comparison, the widely used PMx methods show on average 1.0 kcal mol–1 larger deviations. Furthermore, the application of single-point, multilevel approaches and the parallelism of potential energy surfaces are discussed.
中文翻译:

金属-有机骨架和多孔有机笼中小分子结合的有效计算
气体和燃料的活化,储存和分离与减少温室气体排放,可再生能源的广泛使用以及工业气体的应用密切相关。金属有机骨架(MOF)和多孔有机笼(POC)是一类新兴的结晶多孔材料,在该领域显示出令人鼓舞的特性。然而,由于孔和笼子的巨大尺寸以及通常复杂的结构,其准确的理论描述对现有方法提出了挑战。在这项工作中,通常适用的密度泛函近似(DFA),半经验量子力学(SQM)方法的性能,参照高水平PBE0-D4 / def2-TZVP混合DFT能量测试了用于计算MOF和POC分子切口中各种气体结合能的力场(FFs)。因此,对于温室气体(CO如图2所示,与能源有关的气体(H 2,甲醇和苯)和工业气体(N 2)由有效的构象搜索算法(CREST)确定。通过DFT(B97-3c),半经验(GFN2-xTB)和力场(GFN-FF)方法进一步优化所得结构,以产生结合位点和相应的能量。对于所有测试系统,平均绝对偏差范围为1.1至1.4 kcal mol –1,考虑到的GFN方法的准确度非常接近DFT参考,证明它们可用于有效的结合位点筛选。相比之下,广泛使用的PM x方法平均显示1.0 kcal mol –1更大的偏差。此外,讨论了单点,多级方法的应用和势能面的平行性。
更新日期:2020-12-17
中文翻译:
金属-有机骨架和多孔有机笼中小分子结合的有效计算
气体和燃料的活化,储存和分离与减少温室气体排放,可再生能源的广泛使用以及工业气体的应用密切相关。金属有机骨架(MOF)和多孔有机笼(POC)是一类新兴的结晶多孔材料,在该领域显示出令人鼓舞的特性。然而,由于孔和笼子的巨大尺寸以及通常复杂的结构,其准确的理论描述对现有方法提出了挑战。在这项工作中,通常适用的密度泛函近似(DFA),半经验量子力学(SQM)方法的性能,参照高水平PBE0-D4 / def2-TZVP混合DFT能量测试了用于计算MOF和POC分子切口中各种气体结合能的力场(FFs)。因此,对于温室气体(CO如图2所示,与能源有关的气体(H 2,甲醇和苯)和工业气体(N 2)由有效的构象搜索算法(CREST)确定。通过DFT(B97-3c),半经验(GFN2-xTB)和力场(GFN-FF)方法进一步优化所得结构,以产生结合位点和相应的能量。对于所有测试系统,平均绝对偏差范围为1.1至1.4 kcal mol –1,考虑到的GFN方法的准确度非常接近DFT参考,证明它们可用于有效的结合位点筛选。相比之下,广泛使用的PM x方法平均显示1.0 kcal mol –1更大的偏差。此外,讨论了单点,多级方法的应用和势能面的平行性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号