当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Topological Analysis of Hydroxyquinoline Derivatives Interacting with Aluminum Cations or with an Al(111) Surface
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2020-12-01 , DOI: 10.1021/acs.inorgchem.0c01972 Yann Bulteau 1 , Christine Lepetit 2 , Corinne Lacaze-Dufaure 1
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2020-12-01 , DOI: 10.1021/acs.inorgchem.0c01972 Yann Bulteau 1 , Christine Lepetit 2 , Corinne Lacaze-Dufaure 1
Affiliation
|
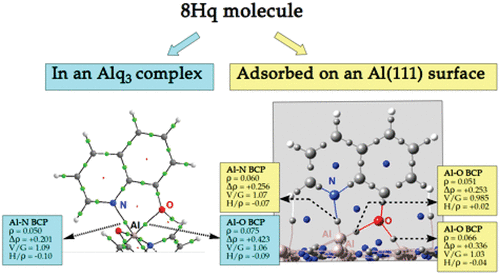
The reactivity of hydroxyquinoline derivatives (native molecules (Hq) and modified species (HqX, X = Br, SO3H, or SO3–)) is investigated either (i) with aluminum cations for the formation of chelates or (ii) with aluminum surfaces for their adsorption properties, in the framework of the dispersion-corrected Density Functional Theory (DFT-D). It is shown that the substituent X has no influence on the complexation to the aluminum cation of the deprotonated active form, i.e., the one exhibiting a phenolate moiety and referred to as q– for the native Hq and qXn– (n = 1 or 2) for its derivatives. The formation energies of the Alq3 and Al(qX)3 complexes, taking values of −60.87 ± 3.10 eV in vacuum and −24.30 ± 0.29 eV in water, are indicative of a strong chelating affinity of the q– and qXn– (n = 1 or 2) anions for the aluminum cations. ELF and QTAIM topological analyses on these complexes evidence that the bonding of the deprotonated species with the Al3+ ion is ionic with a very weak covalence degree. The para or ortho substituent X of the phenolate moiety of the qXn– (n = 1 or 2) derivatives modifies the electronic structure only locally and thus does not influence their O- or N-coordinating properties. The adsorption properties of the latter on an Al(111) surface have also been studied within periodic DFT-D calculations. The adsorbed species are strongly interacting with the Al(111) surface, as shown by the value of the adsorption energy of −3.69 ± 0.21 eV for the most stable geometries. Various adsorption modes of the q– and qXn– (n = 1 or 2) derivatives are characterized on the Al surface, depending on stabilizing or destabilizing interactions with the substituents X. On the basis of QTAIM descriptors, the bonding of the hydroxyquinoline species on the aluminum surface is characterized as ionic with a weak covalent character.
中文翻译:

与铝阳离子或Al(111)表面相互作用的羟基喹啉衍生物的拓扑分析
羟基喹啉衍生物的反应性(天然分子(李国华)和改性物种(HQX,X = Br的,SO 3 H,或SO 3 - ))与铝阳离子任一研究(i)用于螯合物的形成或(ii)与铝表面在分散校正的密度泛函理论(DFT-D)框架中的吸附性能。结果表明,该取代基X具有对络合到去质子化的活性形式,即,一个呈现酚部分和被称为的铝阳离子没有影响q -对天然李国华和QX ñ - (Ñ= 1或2)。Alq 3和Al(qX)3配合物的形成能在真空中为−60.87±3.10 eV,在水中为−24.30±0.29 eV,表明q –和qX n –()具有很强的螯合亲和力。n = 1或2)铝阳离子的阴离子。对这些配合物进行的ELF和QTAIM拓扑分析表明,去质子化物质与Al 3+离子的键合是离子性的,具有很弱的共价度。的对位或邻位取代基的的酚部分的X QX ñ - (n = 1或2)衍生物仅局部改变电子结构,因此不影响其O或N配位性质。在周期性DFT-D计算中还研究了后者在Al(111)表面上的吸附性能。吸附的物质与Al(111)表面强烈相互作用,如最稳定几何形状的吸附能值为-3.69±0.21 eV。q –和qX n –(n的各种吸附模式 = 1或2)衍生物在Al表面上的特征取决于与取代基X的稳定或不稳定相互作用。基于QTAIM描述子,铝表面上羟基喹啉类的键合被表征为具有弱共价特性的离子型。
更新日期:2020-12-21
中文翻译:
与铝阳离子或Al(111)表面相互作用的羟基喹啉衍生物的拓扑分析
羟基喹啉衍生物的反应性(天然分子(李国华)和改性物种(HQX,X = Br的,SO 3 H,或SO 3 - ))与铝阳离子任一研究(i)用于螯合物的形成或(ii)与铝表面在分散校正的密度泛函理论(DFT-D)框架中的吸附性能。结果表明,该取代基X具有对络合到去质子化的活性形式,即,一个呈现酚部分和被称为的铝阳离子没有影响q -对天然李国华和QX ñ - (Ñ= 1或2)。Alq 3和Al(qX)3配合物的形成能在真空中为−60.87±3.10 eV,在水中为−24.30±0.29 eV,表明q –和qX n –()具有很强的螯合亲和力。n = 1或2)铝阳离子的阴离子。对这些配合物进行的ELF和QTAIM拓扑分析表明,去质子化物质与Al 3+离子的键合是离子性的,具有很弱的共价度。的对位或邻位取代基的的酚部分的X QX ñ - (n = 1或2)衍生物仅局部改变电子结构,因此不影响其O或N配位性质。在周期性DFT-D计算中还研究了后者在Al(111)表面上的吸附性能。吸附的物质与Al(111)表面强烈相互作用,如最稳定几何形状的吸附能值为-3.69±0.21 eV。q –和qX n –(n的各种吸附模式 = 1或2)衍生物在Al表面上的特征取决于与取代基X的稳定或不稳定相互作用。基于QTAIM描述子,铝表面上羟基喹啉类的键合被表征为具有弱共价特性的离子型。


























 京公网安备 11010802027423号
京公网安备 11010802027423号