当前位置:
X-MOL 学术
›
Hum. Mutat.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Biallelic TMEM251 variants in patients with severe skeletal dysplasia and extreme short stature
Human Mutation ( IF 3.9 ) Pub Date : 2020-11-30 , DOI: 10.1002/humu.24139 Noor U Ain 1, 2 , Niaz Muhammad 1 , Mehdi Dianatpour 3, 4 , Marta Baroncelli 5 , Muddassar Iqbal 1 , Mohammad A F Fard 6 , Ihtisham Bukhari 1 , Sufian Ahmed 1 , Massoumeh Hajipour 6 , Zahra Tabatabaie 6 , Hamidreza Foroutan 7 , Ola Nilsson 5, 8 , Mohammad A Faghihi 6 , Outi Makitie 2, 9, 10 , Sadaf Naz 1
Human Mutation ( IF 3.9 ) Pub Date : 2020-11-30 , DOI: 10.1002/humu.24139 Noor U Ain 1, 2 , Niaz Muhammad 1 , Mehdi Dianatpour 3, 4 , Marta Baroncelli 5 , Muddassar Iqbal 1 , Mohammad A F Fard 6 , Ihtisham Bukhari 1 , Sufian Ahmed 1 , Massoumeh Hajipour 6 , Zahra Tabatabaie 6 , Hamidreza Foroutan 7 , Ola Nilsson 5, 8 , Mohammad A Faghihi 6 , Outi Makitie 2, 9, 10 , Sadaf Naz 1
Affiliation
|
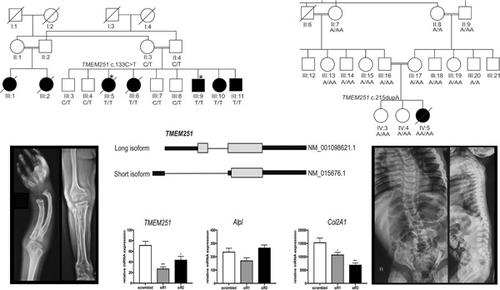
Skeletal dysplasias are a heterogeneous group of disorders ranging from mild to lethal skeletal defects. We investigated two unrelated families with individuals presenting with a severe skeletal disorder. In family NMD02, affected individuals had a dysostosis multiplex‐like skeletal dysplasia and severe short stature (<−8.5 SD). They manifested increasingly coarse facial features, protruding abdomens, and progressive skeletal changes, reminiscent of mucopolysaccharidosis. The patients gradually lost mobility and the two oldest affected individuals died in their twenties. The affected child in family ID01 had coarse facial features and severe skeletal dysplasia with clinical features similar to mucopolysaccharidosis. She had short stature, craniosynostosis, kyphoscoliosis, and hip‐joint subluxation. She died at the age of 5 years. Whole‐exome sequencing identified two homozygous variants c.133C>T; p.(Arg45Trp) and c.215dupA; p.(Tyr72Ter), respectively, in the two families, affecting an evolutionary conserved gene TMEM251 (NM_001098621.1). Immunofluorescence and confocal studies using human osteosarcoma cells indicated that TMEM251 is localized to the Golgi complex. However, p.Arg45Trp mutant TMEM251 protein was targeted less efficiently and the localization was punctate. Tmem251 knockdown by small interfering RNA induced dedifferentiation of rat primary chondrocytes. Our work implicates TMEM251 in the pathogenesis of a novel disorder and suggests its potential function in chondrocyte differentiation.
中文翻译:

严重骨骼发育不良和身材矮小患者的双等位基因 TMEM251 变异
骨骼发育不良是一组异质性疾病,范围从轻度到致命的骨骼缺陷。我们调查了两个不相关的家庭,其中的个人患有严重的骨骼疾病。在 NMD02 家族中,受影响的个体患有多发性骨发育不全样骨骼发育不良和严重身材矮小(<−8.5 SD)。他们表现出越来越粗糙的面部特征、突出的腹部和进行性的骨骼变化,让人想起粘多糖中毒。患者逐渐失去行动能力,其中两名最年长的患者在二十多岁时死亡。ID01家系患儿面部特征粗糙,骨骼严重发育不良,临床特征与粘多糖增多症相似。她身材矮小,患有颅缝早闭、脊柱后侧凸和髋关节半脱位。她5岁时去世。全外显子组测序鉴定出两个纯合变异 c.133C>T;p.(Arg45Trp)和c.215dupA;p.(Tyr72Ter),分别在两个家族中,影响进化保守基因TMEM251 (NM_001098621.1)。使用人骨肉瘤细胞的免疫荧光和共聚焦研究表明 TMEM251 定位于高尔基复合体。然而,p.Arg45Trp 突变体 TMEM251 蛋白的靶向效率较低,且定位呈点状。小干扰 RNA 敲低Tmem251诱导大鼠原代软骨细胞去分化。我们的工作表明TMEM251与一种新型疾病的发病机制有关,并表明其在软骨细胞分化中的潜在功能。
更新日期:2020-12-26
中文翻译:
严重骨骼发育不良和身材矮小患者的双等位基因 TMEM251 变异
骨骼发育不良是一组异质性疾病,范围从轻度到致命的骨骼缺陷。我们调查了两个不相关的家庭,其中的个人患有严重的骨骼疾病。在 NMD02 家族中,受影响的个体患有多发性骨发育不全样骨骼发育不良和严重身材矮小(<−8.5 SD)。他们表现出越来越粗糙的面部特征、突出的腹部和进行性的骨骼变化,让人想起粘多糖中毒。患者逐渐失去行动能力,其中两名最年长的患者在二十多岁时死亡。ID01家系患儿面部特征粗糙,骨骼严重发育不良,临床特征与粘多糖增多症相似。她身材矮小,患有颅缝早闭、脊柱后侧凸和髋关节半脱位。她5岁时去世。全外显子组测序鉴定出两个纯合变异 c.133C>T;p.(Arg45Trp)和c.215dupA;p.(Tyr72Ter),分别在两个家族中,影响进化保守基因TMEM251 (NM_001098621.1)。使用人骨肉瘤细胞的免疫荧光和共聚焦研究表明 TMEM251 定位于高尔基复合体。然而,p.Arg45Trp 突变体 TMEM251 蛋白的靶向效率较低,且定位呈点状。小干扰 RNA 敲低Tmem251诱导大鼠原代软骨细胞去分化。我们的工作表明TMEM251与一种新型疾病的发病机制有关,并表明其在软骨细胞分化中的潜在功能。


























 京公网安备 11010802027423号
京公网安备 11010802027423号