Clinical Genetics ( IF 3.5 ) Pub Date : 2020-11-27 , DOI: 10.1111/cge.13885 Katsuhiko Mineta 1 , Kosuke Goto 1 , Takashi Gojobori 1 , Fowzan S Alkuraya 2, 3
|
To the Editor,
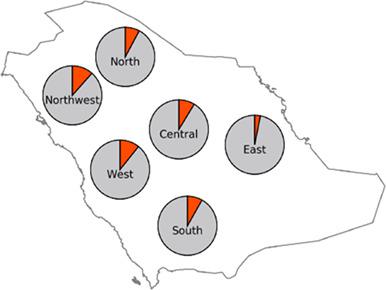
SARS‐CoV‐2 has been identified as the cause of an ongoing pandemic (COVID‐19) that has infected more than 25 m individuals and caused more than 1 m deaths worldwide (WHO). The highly variable clinical course despite a relatively stable viral genome strongly implicates host factors, including genetics. Mendelian large effect variants have recently been identified although these likely account for a very small number of cases.1 On the other hand, the contribution of several common variants has been demonstrated,2 particularly one locus on chr3 which was identified in the first major GWAS on genetic predisposition to severe COVID19.3 Interestingly, a very recent study has convincingly shown that the risk haplotype in the chr3 locus was introgressed into modern humans from Neanderthal.4 The distribution of this risk haplotype was estimated for a wide range of human populations although Middle Eastern Arabs were missing.4 Here, we calculate the distribution of the risk haplotype in Arabia and discuss that in the context of the overall Neanderthal ancestry in the local population. Representative samples from the major indigenous tribes in Arabia were chosen for analysis with informed consent and genotyped as described in detail elsewhere (Mineta et al, 2020).5 We first confirmed by Haploview that the 13 SNPs that constitute the risk haplotype are in complete LD in indigenous Arabs using previously published WGS data. We then tested the frequency of rs13078854 in 953 samples representing the 28 major indigenous tribes in Arabia. The overall risk allele frequency was 8.6% with 135 heterozygotes and 14 homozygotes (of note, homozygotes had only been documented among South Asians [~10%] and 1 individual in Colombia). As shown in Figure 1, the distribution was largely similar between the different regions.

The very recent revelation that the best‐known risk haplotype for COVID‐19 severity is derived from Neanderthal has important evolutionary and clinical implications.4 Most surprisingly, the distribution of this haplotype in the world major populations did not fully correlate with the known distribution of Neanderthal ancestry. While Africans predictably lacked the risk haplotype since they are known to have no significant Neanderthal ancestry, the dramatic enrichment among South Asians and near elimination among East Asians are at odds with published estimates of Neanderthal contribution to these populations. This suggests that this risk allele may have gone through strong selection pressure in opposing directionality in different populations.4 Native inhabitants of Arabia, or indigenous Arabs, have been hypothesized to represent a very ancient split of the early wave of out‐of‐Africa human migration, which is consistent with the rather low Neanderthal contribution of 2.6%.6 It remains unclear at this point if the higher frequency of the risk haplotype (8.6%) represents positive selection since this has not been confirmed even in populations with a much higher frequency, for example, 37.8% in Bangladesh. Clinically, it is worth highlighting that despite the compelling effect of the risk haplotype on the severity of COVID‐19 (OR 2.14),2 the fact that its world distribution has only very recently been established makes it hard to conclude if this correlates with ethnic variation in disease severity. It has been noted, for example, that Bangladeshi patients in the UK are twice as likely to have severe COVID‐19 (Public Health England). However, case fatality rate in Bangladesh remains relatively low at 1.5% (WHO), which is identical to Saudi Arabia (WHO) despite the dramatic difference in the risk haplotype distribution. Clearly, the risk haplotype is but one of many factors that influence the outcome of SARS‐CoV‐2 infection. Nonetheless, it will be helpful to track patient outcome by this haplotype in ethnically diverse cohorts to establish a more general estimate of its clinical impact.
中文翻译:
与世界其他人群相比,土著阿拉伯人具有中等频率的尼安德特人 COVID-19 风险单倍型
致编辑,
SARS-CoV-2 已被确定为持续大流行 (COVID-19) 的原因,该流行病已感染超过 2500 万人,并导致全球超过 100 万人死亡 (WHO)。尽管病毒基因组相对稳定,但临床过程的高度可变性强烈暗示宿主因素,包括遗传学。最近已经确定了孟德尔大效应变体,尽管这些可能占极少数情况。1另一方面,已经证明了几种常见变体的贡献,2特别是 chr3 上的一个基因座,该基因座在关于严重 COVID19 遗传易感性的第一个主要 GWAS 中被确定。3有趣的是,最近的一项研究令人信服地表明,chr3 基因座中的风险单倍型已渗入尼安德特人的现代人类中。4尽管缺少中东阿拉伯人,但对广泛人群的这种风险单倍型分布进行了估计。4在这里,我们计算了阿拉伯地区的风险单倍型分布,并在当地人口的整体尼安德特人血统背景下进行了讨论。在知情同意的情况下选择来自阿拉伯主要土著部落的代表性样本进行分析,并按照其他地方的详细描述进行基因分型(Mineta 等,2020)。5我们首先由 Haploview 证实,使用先前公布的 WGS 数据,构成风险单倍型的 13 个 SNP 在土著阿拉伯人中处于完全 LD 状态。然后,我们在代表阿拉伯 28 个主要土著部落的 953 个样本中测试了 rs13078854 的频率。总体风险等位基因频率为 8.6%,其中 135 个杂合子和 14 个纯合子(值得注意的是,纯合子仅记录在南亚人 [~10%] 和哥伦比亚 1 个个体中)。如图 1 所示,不同地区之间的分布基本相似。
最近发现,最知名的 COVID-19 严重性风险单倍型来自尼安德特人,具有重要的进化和临床意义。4最令人惊讶的是,这种单倍型在世界主要人群中的分布与已知的尼安德特人血统分布并不完全相关。虽然可以预见的是,非洲人缺乏风险单倍型,因为众所周知他们没有显着的尼安德特人血统,但南亚人的显着富集和东亚人的近乎灭绝与已发表的尼安德特人对这些人口的贡献的估计不一致。这表明这种风险等位基因可能在不同人群中经历了相反方向性的强烈选择压力。4阿拉伯原住民或土著阿拉伯人被假设代表了早期非洲人类迁徙浪潮的一个非常古老的分裂,这与尼安德特人 2.6% 的相当低的贡献一致。6目前尚不清楚风险单倍型的较高频率 (8.6%) 是否代表阳性选择,因为即使在频率高得多的人群中也未证实这一点,例如孟加拉国的 37.8%。在临床上,值得强调的是,尽管风险单倍型对 COVID-19 的严重程度有显着影响(OR 2.14),2事实上,它的世界分布是最近才确定的,因此很难断定这是否与疾病严重程度的种族差异有关。例如,已经注意到英国的孟加拉国患者患严重 COVID-19 的可能性是其两倍(英国公共卫生部)。然而,尽管风险单倍型分布存在巨大差异,但孟加拉国的病死率仍然相对较低,为 1.5% (WHO),与沙特阿拉伯 (WHO) 相同。显然,风险单倍型只是影响 SARS-CoV-2 感染结果的众多因素之一。尽管如此,在种族多样化的队列中通过这种单倍型跟踪患者的结果将有助于建立对其临床影响的更一般的估计。

























 京公网安备 11010802027423号
京公网安备 11010802027423号