当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
UV-to-IR Absorption of Molecularly p-Doped Polythiophenes with Alkyl and Oligoether Side Chains: Experiment and Interpretation Based on Density Functional Theory
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-25 , DOI: 10.1021/acs.jpcb.0c08757 Ihor Sahalianov 1 , Jonna Hynynen 2 , Stephen Barlow 3 , Seth R. Marder 3 , Christian Müller 2 , Igor Zozoulenko 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-25 , DOI: 10.1021/acs.jpcb.0c08757 Ihor Sahalianov 1 , Jonna Hynynen 2 , Stephen Barlow 3 , Seth R. Marder 3 , Christian Müller 2 , Igor Zozoulenko 1
Affiliation
|
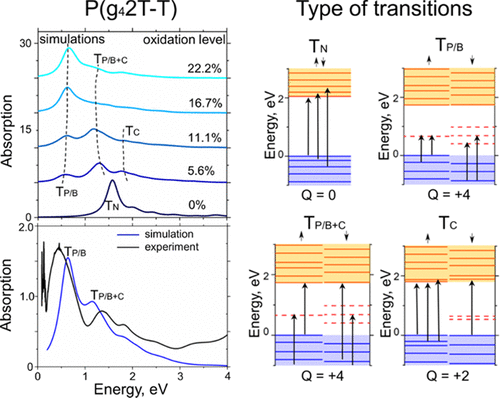
The UV-to-IR transitions in p-doped poly(3-hexylthiophene) (P3HT) with alkyl side chains and polar polythiophene with tetraethylene glycol side chains are studied experimentally by means of the absorption spectroscopy and computationally using density functional theory (DFT) and tight-binding DFT. The evolution of electronic structure is calculated as the doping level is varied, while the roles of dopant ions, chain twisting, and π–π stacking are also considered, each of these having the effect of broadening the absorption peaks while not significantly changing their positions. The calculated spectra are found to be in good agreement with experimental spectra obtained for the polymers doped with a molybdenum dithiolene complex. As in other DFT studies of doped conjugated polymers, the electronic structure and assignment of optical transitions that emerge are qualitatively different from those obtained through earlier “traditional” approaches. In particular, the two prominent bands seen for the p-doped materials are present for both polarons and bipolarons/polaron pairs. The lowest energy of these transitions is due to excitation from the valence band to a spin-resolved orbitals located in the gap between the bands. The higher-energy band is a superposition of excitation from the valence band to a spin-resolved orbitals in the gap and an excitation between bands.
中文翻译:

带有烷基和寡醚侧链的分子对p掺杂聚噻吩的UV-IR吸收:基于密度泛函理论的实验和解释
利用吸收光谱法并通过密度泛函理论(DFT)通过实验研究了具有烷基侧链的p掺杂聚(3-己基噻吩)(P3HT)和具有四乙二醇侧链的极性聚噻吩的紫外-红外跃迁和紧密绑定的DFT。随着掺杂水平的变化,电子结构的演化被计算出来,同时还考虑了掺杂离子,链扭曲和π-π堆叠的作用,这些都具有扩大吸收峰的作用,而不会显着改变其位置。发现计算的光谱与掺杂有二硫代钼钼络合物的聚合物获得的实验光谱非常吻合。与其他对掺杂共轭聚合物的DFT研究一样,电子结构和出现的光学跃迁的分配在质量上与通过早期“传统”方法获得的跃迁不同。特别地,极化子和双极化子/极化子对均存在p掺杂材料的两个突出的带。这些跃迁的最低能量归因于从价带到位于带之间的间隙中的自旋分辨轨道的激发。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。这些跃迁的最低能量归因于从价带激发到位于带之间的间隙中的自旋分辨轨道。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。这些跃迁的最低能量归因于从价带激发到位于带之间的间隙中的自旋分辨轨道。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。
更新日期:2020-12-10
中文翻译:
带有烷基和寡醚侧链的分子对p掺杂聚噻吩的UV-IR吸收:基于密度泛函理论的实验和解释
利用吸收光谱法并通过密度泛函理论(DFT)通过实验研究了具有烷基侧链的p掺杂聚(3-己基噻吩)(P3HT)和具有四乙二醇侧链的极性聚噻吩的紫外-红外跃迁和紧密绑定的DFT。随着掺杂水平的变化,电子结构的演化被计算出来,同时还考虑了掺杂离子,链扭曲和π-π堆叠的作用,这些都具有扩大吸收峰的作用,而不会显着改变其位置。发现计算的光谱与掺杂有二硫代钼钼络合物的聚合物获得的实验光谱非常吻合。与其他对掺杂共轭聚合物的DFT研究一样,电子结构和出现的光学跃迁的分配在质量上与通过早期“传统”方法获得的跃迁不同。特别地,极化子和双极化子/极化子对均存在p掺杂材料的两个突出的带。这些跃迁的最低能量归因于从价带到位于带之间的间隙中的自旋分辨轨道的激发。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。这些跃迁的最低能量归因于从价带激发到位于带之间的间隙中的自旋分辨轨道。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。这些跃迁的最低能量归因于从价带激发到位于带之间的间隙中的自旋分辨轨道。高能带是从价带到间隙中自旋分辨轨道的激发和能带之间的激发的叠加。


























 京公网安备 11010802027423号
京公网安备 11010802027423号