当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
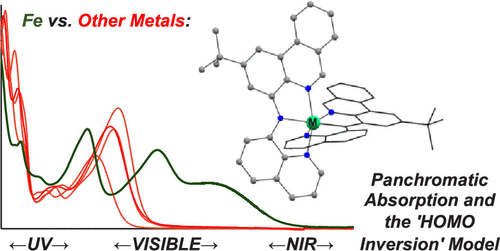
In Pursuit of Panchromatic Absorption in Metal Coordination Complexes: Experimental Delineation of the HOMO Inversion Model Using Pseudo-Octahedral Complexes of Diarylamido Ligands
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.inorgchem.0c02973 Jason D. Braun 1 , Issiah B. Lozada 1 , David E. Herbert 1
Inorganic Chemistry ( IF 4.6 ) Pub Date : 2020-11-23 , DOI: 10.1021/acs.inorgchem.0c02973 Jason D. Braun 1 , Issiah B. Lozada 1 , David E. Herbert 1
Affiliation
|
The ability of a compound to broadly absorb light across the incident solar spectrum is an important design target in the development of molecular photosensitizers. The ‘HOMO inversion’ model predicts that for [(tpy)2Fe]2+ (tpy = 2,2′:6′,2″-terpyridine) compounds, adjusting the character of the highest occupied molecular orbital (HOMO) from metal-centered to ligand-centered can drastically improve photophysical properties by broadening absorption in the visible and increasing molar extinction coefficients. In an effort to experimentally realize strong, panchromatic absorption, a tridentate N^N–^N diarylamido ligand bearing flanking benzannulated N-heterocyclic donors (tBuL) was used to prepare deeply colored, pseudo-octahedral coordination complexes of a range of first-row transition and main-group metals [(tBuL)2M0/+; M = Fe, Co, Ni, Zn, Ga]. While the Fe(II) congener exhibits the sought-after broad absorption, isostructural and isoelectronic complexes of other first-row transition and main-group metals show vastly different absorption and redox properties. Density functional theory (DFT) calculations point toward the relative energies of the metal d orbitals and ligand orbitals as the source of major changes in electronic structure, confirming aspects and limitations of the predictive 'HOMO inversion' model in experimentally realized systems with implications for the design of abundant transition-metal sensitizers with broad, panchromatic absorptive properties.
中文翻译:

追求金属配合物的全色吸收:使用二芳基酰胺配体的伪八面体络合物的HOMO反演模型的实验描述。
化合物在整个入射太阳光谱中广泛吸收光的能力是分子光敏剂开发中的重要设计目标。“ HOMO倒置”模型预测,对于[[tpy)2 Fe] 2+(tpy = 2,2':6',2″-叔吡啶)化合物,可调节金属中最高占据分子轨道(HOMO)的特性中心到配体中心可以通过扩大可见光的吸收并增加摩尔消光系数来显着改善光物理性质。为了通过实验实现强的全色吸收,需要一个三齿N ^ N – ^ N二芳基配体,其侧基为苯环环化的N-杂环供体(t BuL)用于制备一系列第一行过渡金属和主族金属[(t Bu L)2 M 0 / +的深色,伪八面体配位络合物。M = Fe,Co,Ni,Zn,Ga]。尽管Fe(II)同类物表现出广受欢迎的吸收性,但其他第一行过渡金属和主族金属的同构和等电子络合物却表现出截然不同的吸收和氧化还原特性。密度泛函理论(DFT)的计算指向金属d的相对能 轨道和配体轨道是电子结构发生重大变化的根源,这证实了在实验实现的系统中预测性“ HOMO倒置”模型的各个方面和局限性,这对设计具有宽泛,全色吸收特性的过渡金属敏化剂具有重要意义。
更新日期:2020-12-07
中文翻译:
追求金属配合物的全色吸收:使用二芳基酰胺配体的伪八面体络合物的HOMO反演模型的实验描述。
化合物在整个入射太阳光谱中广泛吸收光的能力是分子光敏剂开发中的重要设计目标。“ HOMO倒置”模型预测,对于[[tpy)2 Fe] 2+(tpy = 2,2':6',2″-叔吡啶)化合物,可调节金属中最高占据分子轨道(HOMO)的特性中心到配体中心可以通过扩大可见光的吸收并增加摩尔消光系数来显着改善光物理性质。为了通过实验实现强的全色吸收,需要一个三齿N ^ N – ^ N二芳基配体,其侧基为苯环环化的N-杂环供体(t BuL)用于制备一系列第一行过渡金属和主族金属[(t Bu L)2 M 0 / +的深色,伪八面体配位络合物。M = Fe,Co,Ni,Zn,Ga]。尽管Fe(II)同类物表现出广受欢迎的吸收性,但其他第一行过渡金属和主族金属的同构和等电子络合物却表现出截然不同的吸收和氧化还原特性。密度泛函理论(DFT)的计算指向金属d的相对能 轨道和配体轨道是电子结构发生重大变化的根源,这证实了在实验实现的系统中预测性“ HOMO倒置”模型的各个方面和局限性,这对设计具有宽泛,全色吸收特性的过渡金属敏化剂具有重要意义。


























 京公网安备 11010802027423号
京公网安备 11010802027423号