当前位置:
X-MOL 学术
›
Chemistryopen
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
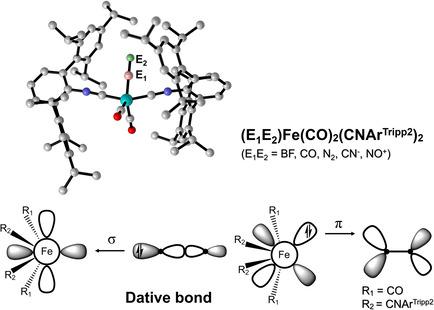
Stabilities, Electronic Structures, and Bonding Properties of Iron Complexes (E1E2)Fe(CO)2(CNArTripp2)2 (E1E2=BF, CO, N2, CN−, or NO+)**
ChemistryOpen ( IF 2.3 ) Pub Date : 2020-11-18 , DOI: 10.1002/open.202000248 Gerui Pei 1 , Pei Zhao 2, 3 , Song Xu 1 , Xintian Zhao 1 , Chuncai Kong 1 , Zhimao Yang 1 , Masahiro Ehara 2, 3 , Tao Yang 1
ChemistryOpen ( IF 2.3 ) Pub Date : 2020-11-18 , DOI: 10.1002/open.202000248 Gerui Pei 1 , Pei Zhao 2, 3 , Song Xu 1 , Xintian Zhao 1 , Chuncai Kong 1 , Zhimao Yang 1 , Masahiro Ehara 2, 3 , Tao Yang 1
Affiliation
|
The coordination of 10‐electron diatomic ligands (BF, CO N2) to iron complexes Fe(CO)2(CNArTripp2)2 [ArTripp2=2,6‐(2,4,6‐(iso‐propyl)3C6H2)2C6H3] have been realized in experiments very recently (Science, 2019, 363, 1203–1205). Herein, the stability, electronic structures, and bonding properties of (E1E2)Fe‐(CO)2(CNArTripp2)2 (E1E2=BF, CO, N2, CN−, NO+) were studied using density functional (DFT) calculations. The ground state of all those molecules is singlet and the calculated geometries are in excellent agreement with the experimental values. The natural bond orbital analysis revealed that Fe is negatively charged while E1 possesses positive charges. By employing the energy decomposition analysis, the bonding nature of the E2E1–Fe(CO)2(CNArTripp2)2 bond was disclosed to be the classic dative bond E2E1→Fe(CO)2(CNArTripp2)2 rather than the electron‐sharing double bond. More interestingly, the bonding strength between BF and Fe(CO)2(CNArTripp2)2 is much stronger than that between CO (or N2) and Fe(CO)2(CNArTripp2)2, which is ascribed to the better σ‐donation and π back‐donations. However, the orbital interactions in CN−→Fe(CO)2(CNArTripp2)2 and NO+→Fe(CO)2(CNArTripp2)2 mainly come from σ‐donation and π back‐donation, respectively. The different contributions from σ donation and π donation for different ligands can be well explained by using the energy levels of E1E2 and Fe(CO)2(CNArTripp2)2 fragments.
中文翻译:

铁配合物 (E1E2)Fe(CO)2(CNArTripp2)2 (E1E2=BF, CO, N2, CN−, or NO+) 的稳定性、电子结构和键合性能**
10-电子双原子配体 (BF, CO N 2 ) 与铁配合物 Fe(CO) 2 (CNAr Tripp2 ) 2 [Ar Tripp2 =2,6-(2,4,6-(异丙基) 3 C ) 的配位6 H 2 ) 2 C 6 H 3 ] 最近在实验中实现了 ( Science , 2019 , 363 , 1203–1205)。在此,(E 1 E 2 )Fe-(CO) 2 (CNAr Tripp2 ) 2的稳定性、电子结构和键合性能(E 1 E 2 =BF、CO、N 2、CN -、NO + )使用密度泛函(DFT)计算进行了研究。所有这些分子的基态都是单线态的,计算出的几何形状与实验值非常吻合。自然键轨道分析表明,Fe 带负电荷,而 E 1带正电荷。通过能量分解分析,E 2 E 1 -Fe(CO) 2 (CNAr Tripp2 ) 2键的键合性质被揭示为经典的配位键 E 2 E 1 →Fe(CO) 2(CNAr Tripp2 ) 2而不是电子共享双键。更有趣的是,BF 和 Fe(CO) 2 (CNAr Tripp2 ) 2之间的结合强度比 CO(或 N 2)和 Fe(CO) 2 (CNAr Tripp2 ) 2之间的结合强度要强得多,这归因于更好的 σ -捐赠和π回馈。然而,CN - →Fe(CO) 2 (CNAr Tripp2 ) 2和 NO + →Fe(CO) 2 (CNAr Tripp2 ) 2中的轨道相互作用主要来自 σ-donation 和 π back-donation。通过使用 E 1 E 2和 Fe(CO) 2 (CNAr Tripp2 ) 2片段的能级可以很好地解释不同配体的 σ 捐赠和 π 捐赠的不同贡献。
更新日期:2020-11-18
中文翻译:
铁配合物 (E1E2)Fe(CO)2(CNArTripp2)2 (E1E2=BF, CO, N2, CN−, or NO+) 的稳定性、电子结构和键合性能**
10-电子双原子配体 (BF, CO N 2 ) 与铁配合物 Fe(CO) 2 (CNAr Tripp2 ) 2 [Ar Tripp2 =2,6-(2,4,6-(异丙基) 3 C ) 的配位6 H 2 ) 2 C 6 H 3 ] 最近在实验中实现了 ( Science , 2019 , 363 , 1203–1205)。在此,(E 1 E 2 )Fe-(CO) 2 (CNAr Tripp2 ) 2的稳定性、电子结构和键合性能(E 1 E 2 =BF、CO、N 2、CN -、NO + )使用密度泛函(DFT)计算进行了研究。所有这些分子的基态都是单线态的,计算出的几何形状与实验值非常吻合。自然键轨道分析表明,Fe 带负电荷,而 E 1带正电荷。通过能量分解分析,E 2 E 1 -Fe(CO) 2 (CNAr Tripp2 ) 2键的键合性质被揭示为经典的配位键 E 2 E 1 →Fe(CO) 2(CNAr Tripp2 ) 2而不是电子共享双键。更有趣的是,BF 和 Fe(CO) 2 (CNAr Tripp2 ) 2之间的结合强度比 CO(或 N 2)和 Fe(CO) 2 (CNAr Tripp2 ) 2之间的结合强度要强得多,这归因于更好的 σ -捐赠和π回馈。然而,CN - →Fe(CO) 2 (CNAr Tripp2 ) 2和 NO + →Fe(CO) 2 (CNAr Tripp2 ) 2中的轨道相互作用主要来自 σ-donation 和 π back-donation。通过使用 E 1 E 2和 Fe(CO) 2 (CNAr Tripp2 ) 2片段的能级可以很好地解释不同配体的 σ 捐赠和 π 捐赠的不同贡献。


























 京公网安备 11010802027423号
京公网安备 11010802027423号