当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Cohesiveness and Nondiffusive Rotational Jump Dynamics of Protic Ionic Liquid from Dispersion-Corrected FPMD Simulations
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-16 , DOI: 10.1021/acs.jpcb.0c05866 Aritri Biswas 1 , Sathish Dasari 1 , Bhabani S. Mallik 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-11-16 , DOI: 10.1021/acs.jpcb.0c05866 Aritri Biswas 1 , Sathish Dasari 1 , Bhabani S. Mallik 1
Affiliation
|
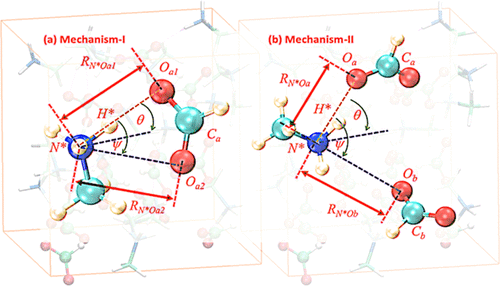
We have investigated the liquid phase of an ionic liquid (IL), methylammonium formate (MAF), through the first principles molecular dynamics simulations using van der Waals (vdW) corrected exchange and correlation functionals of the density functional theory. The simulations were carried out to obtain a comparative study of various properties of the MAF using two different generalized gradient approximation functionals (Becke–Lee–Yang–Parr (BLYP) and Perdew–Burke–Ernzerhof (PBE)) along with three types of dispersion corrections (D2, D3, and dispersion-corrected atom-centered one-electron potentials), and two values of the plane-wave cutoff (300 and 600 Ry). We have evaluated the effects of various electronic parameters in describing the hydrogen-bonded structure and dynamical properties of MAF by performing 10 sets of molecular dynamics simulations. Thermodynamic properties are found to be sensitive to the details of electronic structure calculations. Our results of PBE functionals with the semiempirical vdW method provide the best agreement with experimental density. The overall density predictions match the cohesive energy trends, and the calculations incorporating dispersion forces exhibit enhanced intermolecular interactions within the hydrogen-bonded IL framework. All of the vdW-corrected BLYP functionals, mainly the dispersion-corrected atom-centered one-electron potential (DCACP) method, illustrate a well-defined structure of liquid MAF. To look into the dynamical perspective of the hydrogen-bond descriptions, we elucidate two possible mechanistic pathways of the hydrogen-bond jump events between the counterions. The hydrogen-bond breaking and forming mechanism along with the collision dynamics can be best described by incorporating dispersion interactions alongside the exchange and correlation functionals within the Kohn–Sham scheme. The rattling dynamics of ions are observed for dispersion-corrected functionals. Hence, an accurate representation of the delicately balanced interactive forces within ionic liquids is a necessary step toward a better description of its thermophysical and structural properties along with the associated ionic dynamics.
中文翻译:

色散校正FPMD模拟的质子离子液体的内聚性和非扩散旋转跳跃动力学
我们已经通过使用范德华(vdW)校正的密度泛函理论的交换和相关泛函的第一原理进行了分子动力学模拟,研究了离子液体(IL)的甲酸甲酯甲酸甲酯(MAF)的液相。使用两种不同的色散进行了模拟,以使用两种不同的广义梯度逼近函数(Becke-Lee-Yang-Parr(BLYP)和Perdew-Burke-Ernzerhof(PBE))对MAF的各种特性进行比较研究。校正(D2,D3和色散校正的以原子为中心的单电子电位),以及两个平面波截止值(300和600 Ry)。我们通过执行10组分子动力学模拟,评估了各种电子参数对描述MAF的氢键结构和动力学性质的影响。发现热力学性质对电子结构计算的细节敏感。我们用半经验vdW方法得到的PBE功能的结果与实验密度提供了最好的一致性。总体密度预测与内聚能趋势匹配,并且结合了分散力的计算显示出氢键IL骨架内分子间的相互作用增强。所有vdW校正的BLYP官能团(主要是色散校正的原子中心单电子势(DCACP)方法)都说明了液态MAF的明确结构。为了研究氢键描述的动力学观点,我们阐明了平衡离子之间氢键跳跃事件的两种可能的机理途径。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。
更新日期:2020-11-25
中文翻译:
色散校正FPMD模拟的质子离子液体的内聚性和非扩散旋转跳跃动力学
我们已经通过使用范德华(vdW)校正的密度泛函理论的交换和相关泛函的第一原理进行了分子动力学模拟,研究了离子液体(IL)的甲酸甲酯甲酸甲酯(MAF)的液相。使用两种不同的色散进行了模拟,以使用两种不同的广义梯度逼近函数(Becke-Lee-Yang-Parr(BLYP)和Perdew-Burke-Ernzerhof(PBE))对MAF的各种特性进行比较研究。校正(D2,D3和色散校正的以原子为中心的单电子电位),以及两个平面波截止值(300和600 Ry)。我们通过执行10组分子动力学模拟,评估了各种电子参数对描述MAF的氢键结构和动力学性质的影响。发现热力学性质对电子结构计算的细节敏感。我们用半经验vdW方法得到的PBE功能的结果与实验密度提供了最好的一致性。总体密度预测与内聚能趋势匹配,并且结合了分散力的计算显示出氢键IL骨架内分子间的相互作用增强。所有vdW校正的BLYP官能团(主要是色散校正的原子中心单电子势(DCACP)方法)都说明了液态MAF的明确结构。为了研究氢键描述的动力学观点,我们阐明了平衡离子之间氢键跳跃事件的两种可能的机理途径。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。氢键断裂和形成机理以及碰撞动力学可以通过在Kohn-Sham方案中将色散相互作用与交换和相关功能结合在一起来最好地描述。对于分散校正的功能,观察到了离子的嘎嘎声动力学。因此,准确表示离子液体中微妙的平衡相互作用力是朝更好地描述其热物理和结构特性以及相关离子动力学的必要步骤。
























 京公网安备 11010802027423号
京公网安备 11010802027423号