当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2020-11-16 , DOI: 10.1021/acs.jproteome.0c00563 Ramin Rad 1 , Jiaming Li 1 , Julian Mintseris 1 , Jeremy O’Connell 1 , Steven P. Gygi 1 , Devin K. Schweppe 2
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2020-11-16 , DOI: 10.1021/acs.jproteome.0c00563 Ramin Rad 1 , Jiaming Li 1 , Julian Mintseris 1 , Jeremy O’Connell 1 , Steven P. Gygi 1 , Devin K. Schweppe 2
Affiliation
|
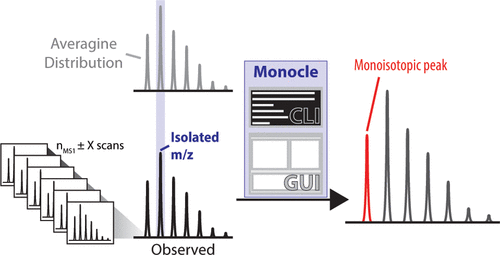
Accurate assignment of monoisotopic peaks is essential for the identification of peptides in bottom-up proteomics. Misassignment or inaccurate attribution of peptidic ions leads to lower sensitivity and fewer total peptide identifications. In the present work, we present a performant, open-source, cross-platform algorithm, Monocle, for the rapid reassignment of instrument-assigned precursor peaks to monoisotopic peptide assignments. We demonstrate that the present algorithm can be integrated into many common proteomic pipelines and provides rapid conversion from multiple data source types. Finally, we show that our monoisotopic peak assignment results in up to a twofold increase in total peptide identifications compared to analyses lacking monoisotopic correction and a 44% improvement over previous monoisotopic peak correction algorithms.
中文翻译:

改进的单同位素质量估计,可实现更深的蛋白质组覆盖
单同位素峰的准确分配对于自下而上的蛋白质组学中肽段的鉴定至关重要。肽离子的错误分配或不正确的归因会导致较低的灵敏度和较少的总肽鉴定。在当前的工作中,我们提出了一种高性能,开放源代码,跨平台的算法Monocle,用于将仪器分配的前体峰快速重新分配给单同位素肽分配。我们证明了本算法可以集成到许多常见的蛋白质组学流水线中,并提供从多种数据源类型的快速转换。最后,我们表明,与缺乏单同位素校正的分析相比,与以往的单同位素峰校正算法相比,我们的单同位素峰分配导致总肽段鉴定最多增加了两倍。
更新日期:2021-01-01
中文翻译:
改进的单同位素质量估计,可实现更深的蛋白质组覆盖
单同位素峰的准确分配对于自下而上的蛋白质组学中肽段的鉴定至关重要。肽离子的错误分配或不正确的归因会导致较低的灵敏度和较少的总肽鉴定。在当前的工作中,我们提出了一种高性能,开放源代码,跨平台的算法Monocle,用于将仪器分配的前体峰快速重新分配给单同位素肽分配。我们证明了本算法可以集成到许多常见的蛋白质组学流水线中,并提供从多种数据源类型的快速转换。最后,我们表明,与缺乏单同位素校正的分析相比,与以往的单同位素峰校正算法相比,我们的单同位素峰分配导致总肽段鉴定最多增加了两倍。

























 京公网安备 11010802027423号
京公网安备 11010802027423号