当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine Learning Prediction of Nine Molecular Properties Based on the SMILES Representation of the QM9 Quantum-Chemistry Dataset
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-11-11 , DOI: 10.1021/acs.jpca.0c05969 Gabriel A. Pinheiro 1 , Johnatan Mucelini 2 , Marinalva D. Soares 3 , Ronaldo C. Prati 4 , Juarez L. F. Da Silva 2 , Marcos G. Quiles 5
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-11-11 , DOI: 10.1021/acs.jpca.0c05969 Gabriel A. Pinheiro 1 , Johnatan Mucelini 2 , Marinalva D. Soares 3 , Ronaldo C. Prati 4 , Juarez L. F. Da Silva 2 , Marcos G. Quiles 5
Affiliation
|
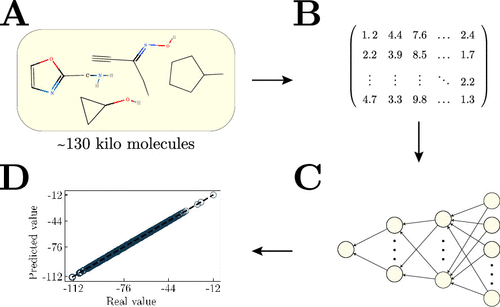
Machine learning (ML) models can potentially accelerate the discovery of tailored materials by learning a function that maps chemical compounds into their respective target properties. In this realm, a crucial step is encoding the molecular systems into the ML model, in which the molecular representation plays a crucial role. Most of the representations are based on the use of atomic coordinates (structure); however, it can increase ML training and predictions’ computational cost. Herein, we investigate the impact of choosing free-coordinate descriptors based on the Simplified Molecular Input Line Entry System (SMILES) representation, which can substantially reduce the ML predictions’ computational cost. Therefore, we evaluate a feed-forward neural network (FNN) model’s prediction performance over five feature selection methods and nine ground-state properties (including energetic, electronic, and thermodynamic properties) from a public data set composed of ∼130k organic molecules. Our best results reached a mean absolute error, close to chemical accuracy, of ∼0.05 eV for the atomization energies (internal energy at 0 K, internal energy at 298.15 K, enthalpy at 298.15 K, and free energy at 298.15 K). Moreover, for the atomization energies, the results obtained an out-of-sample error nine times less than the same FNN model trained with the Coulomb matrix, a traditional coordinate-based descriptor. Furthermore, our results showed how limited the model’s accuracy is by employing such low computational cost representation that carries less information about the molecular structure than the most state-of-the-art methods.
中文翻译:

基于QM9量子化学数据集的SMILES表示的九种分子特性的机器学习预测
机器学习(ML)模型可以通过学习将化合物映射到其各自目标特性的函数来潜在地加速定制材料的发现。在这个领域中,关键的一步是将分子系统编码为ML模型,其中分子表示起着至关重要的作用。大多数表示是基于原子坐标(结构)的使用。但是,它会增加机器学习训练和预测的计算成本。在本文中,我们研究了基于简化分子输入线输入系统(SMILES)表示选择自由坐标描述符的影响,这可以大大降低ML预测的计算成本。因此,我们评估了前馈神经网络(FNN)模型在五种特征选择方法和九种基态性质(包括能量,电子和热力学性质)的预测性能,这些特征集包含约13万个有机分子。我们的最佳结果是,雾化能量(0 K时的内部能量,298.15 K时的内部能量,298.15 K时的焓和298.15 K时的自由能)的平均绝对误差接近于化学精度,约为0.05 eV。此外,对于雾化能量,结果获得的样本外误差比使用传统基于坐标的描述符库仑矩阵训练的相同FNN模型小九倍。此外,
更新日期:2020-11-25
中文翻译:
基于QM9量子化学数据集的SMILES表示的九种分子特性的机器学习预测
机器学习(ML)模型可以通过学习将化合物映射到其各自目标特性的函数来潜在地加速定制材料的发现。在这个领域中,关键的一步是将分子系统编码为ML模型,其中分子表示起着至关重要的作用。大多数表示是基于原子坐标(结构)的使用。但是,它会增加机器学习训练和预测的计算成本。在本文中,我们研究了基于简化分子输入线输入系统(SMILES)表示选择自由坐标描述符的影响,这可以大大降低ML预测的计算成本。因此,我们评估了前馈神经网络(FNN)模型在五种特征选择方法和九种基态性质(包括能量,电子和热力学性质)的预测性能,这些特征集包含约13万个有机分子。我们的最佳结果是,雾化能量(0 K时的内部能量,298.15 K时的内部能量,298.15 K时的焓和298.15 K时的自由能)的平均绝对误差接近于化学精度,约为0.05 eV。此外,对于雾化能量,结果获得的样本外误差比使用传统基于坐标的描述符库仑矩阵训练的相同FNN模型小九倍。此外,


























 京公网安备 11010802027423号
京公网安备 11010802027423号