当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Interaction potential function for the deformation analysis of potassium dihydrogen phosphate using molecular dynamics simulation
Computational Materials Science ( IF 3.3 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110122 Shengyao Yang , Liangchi Zhang , Hongtao Xie , Weidong Liu
Computational Materials Science ( IF 3.3 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.commatsci.2020.110122 Shengyao Yang , Liangchi Zhang , Hongtao Xie , Weidong Liu
|
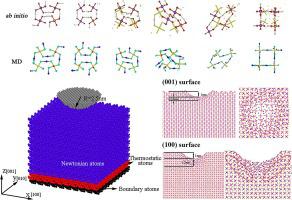
Abstract Potassium dihydrogen phosphate (KDP) is an important nonlinear optical material which plays a core role in electro-optic switches and laser spectroscopy. However, KDP is also one of the most difficult-to-handle materials due to its fragility, unstable microstructure and complex mechanical behaviour. Molecular dynamics (MD) simulation is an appropriate method to explore the deformation mechanisms of the material at the atomic scale. However, the challenge is that there is not a suitable potential function for describing the mechanical behaviour of KDP by using MD simulation. This paper successfully developed a potential function, which enables such insightful investigations. It was found that the established potential function can reliably predict the mechanical properties of KDP including its modulus in different crystal directions and structural changes under various loading conditions.
中文翻译:

使用分子动力学模拟磷酸二氢钾变形分析的相互作用势函数
摘要 磷酸二氢钾(KDP)是一种重要的非线性光学材料,在电光开关和激光光谱中起着核心作用。然而,由于其脆性、不稳定的微观结构和复杂的机械行为,KDP 也是最难处理的材料之一。分子动力学 (MD) 模拟是在原子尺度上探索材料变形机制的合适方法。然而,挑战在于没有合适的势函数来使用 MD 模拟来描述 KDP 的机械行为。这篇论文成功地开发了一个潜在的功能,它能够进行这种有见地的调查。
更新日期:2021-02-01
中文翻译:
使用分子动力学模拟磷酸二氢钾变形分析的相互作用势函数
摘要 磷酸二氢钾(KDP)是一种重要的非线性光学材料,在电光开关和激光光谱中起着核心作用。然而,由于其脆性、不稳定的微观结构和复杂的机械行为,KDP 也是最难处理的材料之一。分子动力学 (MD) 模拟是在原子尺度上探索材料变形机制的合适方法。然而,挑战在于没有合适的势函数来使用 MD 模拟来描述 KDP 的机械行为。这篇论文成功地开发了一个潜在的功能,它能够进行这种有见地的调查。

























 京公网安备 11010802027423号
京公网安备 11010802027423号