当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Thermodynamic Preference for Atom Adsorption on versus Intercalation into Multilayer Graphene
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2020-11-02 , DOI: 10.1021/acs.jpclett.0c02887 Wei Li 1 , Li Huang 1 , Michael C. Tringides 2, 3 , James W. Evans 2, 3 , Yong Han 2, 3
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2020-11-02 , DOI: 10.1021/acs.jpclett.0c02887 Wei Li 1 , Li Huang 1 , Michael C. Tringides 2, 3 , James W. Evans 2, 3 , Yong Han 2, 3
Affiliation
|
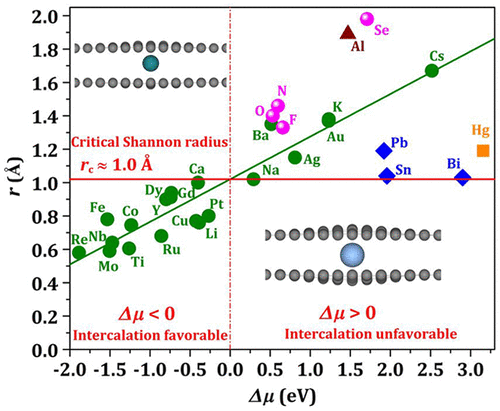
The thermodynamic preference of a foreign atom for adsorption on versus intercalation into a graphitic surface is of fundamental and widespread interest. From an exhaustive first-principles density functional theory investigation for 38 typical elements over the periodic table, we reveal a quasilinear correlation between the Shannon effective ionic radius and the chemical-potential difference for a single atom from adsorption to intercalation at multilayer graphene surfaces. A critical Shannon radius is found to be around 0.10 nm, below (above) which intercalation (adsorption) is more favorable for elements with ionic-like bonding after intercalation. Single atoms with van der Waals-biased bonding show some deviation from the linear relationship, while single atoms for the elements with covalent-like bonding do not favor intercalation relative to adsorption. An energy decomposition analysis indicates that the chemical-potential difference determining the thermodynamic preference of a foreign atom for adsorption versus intercalation results from the competition between the electronic and elastic strain effects.
中文翻译:

吸附原子对多层石墨烯的热力学偏好
基本的和广泛的兴趣是,外来原子的热力学偏好是吸附在石墨表面或相对于插入石墨表面。通过对元素周期表上38个典型元素的详尽的第一原理密度泛函理论研究,我们揭示了香农有效离子半径与单个原子从多层石墨烯表面吸附到嵌入的化学势差之间的拟线性关系。发现临界香农半径为约0.10nm,低于(之上),该嵌入(吸附)对于嵌入后具有离子样键合的元素更有利。具有范德华偏压键的单原子显示出与线性关系的某些偏离,而具有共价键的元素的单原子相对于吸附而言则不利于插层。能量分解分析表明,化学势差决定了外来原子对吸附和插层的热力学偏好,这是由电子和弹性应变效应之间的竞争引起的。
更新日期:2020-11-19
中文翻译:
吸附原子对多层石墨烯的热力学偏好
基本的和广泛的兴趣是,外来原子的热力学偏好是吸附在石墨表面或相对于插入石墨表面。通过对元素周期表上38个典型元素的详尽的第一原理密度泛函理论研究,我们揭示了香农有效离子半径与单个原子从多层石墨烯表面吸附到嵌入的化学势差之间的拟线性关系。发现临界香农半径为约0.10nm,低于(之上),该嵌入(吸附)对于嵌入后具有离子样键合的元素更有利。具有范德华偏压键的单原子显示出与线性关系的某些偏离,而具有共价键的元素的单原子相对于吸附而言则不利于插层。能量分解分析表明,化学势差决定了外来原子对吸附和插层的热力学偏好,这是由电子和弹性应变效应之间的竞争引起的。

























 京公网安备 11010802027423号
京公网安备 11010802027423号