Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Proton extrusion during oxidative burst in microglia exacerbates pathological acidosis following traumatic brain injury
Glia ( IF 6.2 ) Pub Date : 2020-10-22 , DOI: 10.1002/glia.23926 Rodney M Ritzel 1 , Junyun He 1 , Yun Li 1 , Tuoxin Cao 1 , Niaz Khan 1 , Bosung Shim 1 , Boris Sabirzhanov 1 , Taryn Aubrecht 1 , Bogdan A Stoica 1 , Alan I Faden 1, 2 , Long-Jun Wu 3 , Junfang Wu 1, 2
Glia ( IF 6.2 ) Pub Date : 2020-10-22 , DOI: 10.1002/glia.23926 Rodney M Ritzel 1 , Junyun He 1 , Yun Li 1 , Tuoxin Cao 1 , Niaz Khan 1 , Bosung Shim 1 , Boris Sabirzhanov 1 , Taryn Aubrecht 1 , Bogdan A Stoica 1 , Alan I Faden 1, 2 , Long-Jun Wu 3 , Junfang Wu 1, 2
Affiliation
|
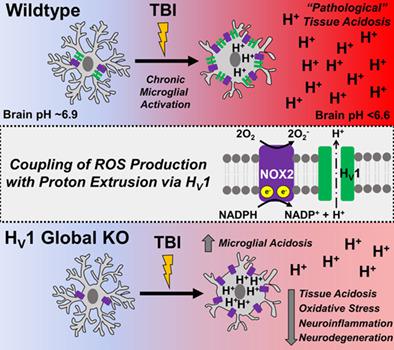
Acidosis is among the least studied secondary injury mechanisms associated with neurotrauma. Acute decreases in brain pH correlate with poor long‐term outcome in patients with traumatic brain injury (TBI), however, the temporal dynamics and underlying mechanisms are unclear. As key drivers of neuroinflammation, we hypothesized that microglia directly regulate acidosis after TBI, and thereby, worsen neurological outcomes. Using a controlled cortical impact model in adult male mice we demonstrate that intracellular pH in microglia and extracellular pH surrounding the lesion site are significantly reduced for weeks after injury. Microglia proliferation and production of reactive oxygen species (ROS) were also increased during the first week, mirroring the increase in extracellular ROS levels seen around the lesion site. Microglia depletion by a colony stimulating factor 1 receptor (CSF1R) inhibitor, PLX5622, markedly decreased extracellular acidosis, ROS production, and inflammation in the brain after injury. Mechanistically, we identified that the voltage‐gated proton channel Hv1 promotes oxidative burst activity and acid extrusion in microglia. Compared to wildtype controls, microglia lacking Hv1 showed reduced ability to generate ROS and extrude protons. Importantly, Hv1‐deficient mice exhibited reduced pathological acidosis and inflammation after TBI, leading to long‐term neuroprotection and functional recovery. Our data therefore establish the microglial Hv1 proton channel as an important link that integrates inflammation and acidosis within the injury microenvironment during head injury.
中文翻译:

小胶质细胞氧化爆发期间的质子挤出加剧了创伤性脑损伤后的病理性酸中毒
酸中毒是研究最少的与神经外伤相关的继发性损伤机制之一。脑 pH 值的急性下降与创伤性脑损伤 (TBI) 患者的长期不良预后相关,然而,时间动态和潜在机制尚不清楚。作为神经炎症的关键驱动因素,我们假设小胶质细胞直接调节 TBI 后的酸中毒,从而恶化神经系统结果。在成年雄性小鼠中使用受控皮质冲击模型,我们证明小胶质细胞中的细胞内 pH 值和病变部位周围的细胞外 pH 值在损伤后数周内显着降低。在第一周,小胶质细胞的增殖和活性氧 (ROS) 的产生也增加,反映了病变部位周围细胞外 ROS 水平的增加。集落刺激因子 1 受体 (CSF1R) 抑制剂 PLX5622 对小胶质细胞的消耗显着降低了损伤后脑内的细胞外酸中毒、ROS 产生和炎症。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。损伤后脑中的细胞外酸中毒、活性氧生成和炎症显着降低。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。损伤后脑中的细胞外酸中毒、活性氧生成和炎症显着降低。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。
更新日期:2020-10-22
中文翻译:
小胶质细胞氧化爆发期间的质子挤出加剧了创伤性脑损伤后的病理性酸中毒
酸中毒是研究最少的与神经外伤相关的继发性损伤机制之一。脑 pH 值的急性下降与创伤性脑损伤 (TBI) 患者的长期不良预后相关,然而,时间动态和潜在机制尚不清楚。作为神经炎症的关键驱动因素,我们假设小胶质细胞直接调节 TBI 后的酸中毒,从而恶化神经系统结果。在成年雄性小鼠中使用受控皮质冲击模型,我们证明小胶质细胞中的细胞内 pH 值和病变部位周围的细胞外 pH 值在损伤后数周内显着降低。在第一周,小胶质细胞的增殖和活性氧 (ROS) 的产生也增加,反映了病变部位周围细胞外 ROS 水平的增加。集落刺激因子 1 受体 (CSF1R) 抑制剂 PLX5622 对小胶质细胞的消耗显着降低了损伤后脑内的细胞外酸中毒、ROS 产生和炎症。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。损伤后脑中的细胞外酸中毒、活性氧生成和炎症显着降低。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。损伤后脑中的细胞外酸中毒、活性氧生成和炎症显着降低。从机制上讲,我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。我们发现电压门控质子通道 Hv1 促进了小胶质细胞的氧化爆发活性和酸排出。与野生型对照相比,缺乏 Hv1 的小胶质细胞产生 ROS 和挤出质子的能力降低。重要的是,Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。Hv1 缺陷小鼠在 TBI 后表现出减少的病理性酸中毒和炎症,从而导致长期的神经保护和功能恢复。因此,我们的数据将小胶质细胞 Hv1 质子通道确立为在头部损伤期间将炎症和酸中毒整合到损伤微环境中的重要环节。

























 京公网安备 11010802027423号
京公网安备 11010802027423号