当前位置:
X-MOL 学术
›
Eur. J. Nerosci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Inflammatory pain in peripheral tissue depends on the activation of the TNF‐α type 1 receptor in the primary afferent neuron
European Journal of Neroscience ( IF 3.4 ) Pub Date : 2020-09-26 , DOI: 10.1111/ejn.14985 Silviane F de Magalhães 1 , Luis P Manzo 1 , Felipe M de Faria 1 , Maria C de Oliveira-Fusaro 2 , Catarine M Nishijima 1 , Willians F Vieira 1 , Ivan J M Bonet 1 , Gilson G Dos Santos 1 , Claudia H Tambeli 1 , Carlos A Parada 1
European Journal of Neroscience ( IF 3.4 ) Pub Date : 2020-09-26 , DOI: 10.1111/ejn.14985 Silviane F de Magalhães 1 , Luis P Manzo 1 , Felipe M de Faria 1 , Maria C de Oliveira-Fusaro 2 , Catarine M Nishijima 1 , Willians F Vieira 1 , Ivan J M Bonet 1 , Gilson G Dos Santos 1 , Claudia H Tambeli 1 , Carlos A Parada 1
Affiliation
|
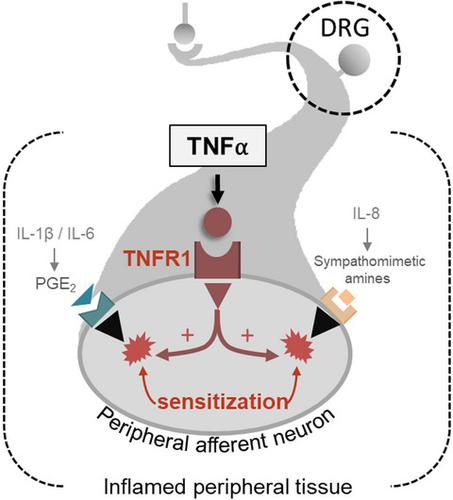
The mechanism underlying the role of tumor necrosis factor alpha (TNF‐α) in the development of inflammatory hyperalgesia has been extensively studied, mainly the role of TNF‐α in the release of pro‐inflammatory cytokines. The current concept relies in the fact that TNF‐α stimulates the cascade release of other pro‐inflammatory cytokines, such as IL‐1β, IL‐6, and IL‐8 (CINC‐1 in rats), triggering the release of the final inflammatory mediator prostaglandin E2 (PGE2) and sympathetic amines that directly sensitize the nociceptors. However, this may not be the sole mechanism involved as the blockade of TNF‐α synthesis by thalidomide prevents hyperalgesia without interrupting the synthesis of IL‐1β, IL‐6, and CINC‐1. Therefore, we hypothesized that activation of TNF‐α receptor type 1 (TNFR1) by TNF‐α increases nociceptors’ susceptibility to the action of PGE2 and dopamine. We have found out that intrathecal administration of oligodeoxynucleotide‐antisense (ODN‐AS) against TNFR1 or thalidomide prevented carrageenan‐induced hyperalgesia. The co‐administration of TNF‐α with a subthreshold dose of PGE2 or dopamine that does not induce hyperalgesia by itself in the hind paw of Wistar rats pretreated with dexamethasone (to prevent the endogenous release of cytokines) induced a robust hyperalgesia that was prevented by intrathecal treatment with ODN‐AS against TNFR1. We consider that the activation of neuronal TNFR1 by TNF‐α decisively increases the susceptibility of the peripheral afferent neuron to the action of final inflammatory mediators – PGE2 and dopamine – that ultimately induce hyperalgesia. This mechanism may also underlie the analgesic action of thalidomide.
中文翻译:

外周组织的炎症性疼痛取决于初级传入神经元中 TNF-α 1 型受体的激活
肿瘤坏死因子α(TNF-α)在炎症性痛觉过敏发展中的作用机制已被广泛研究,主要是TNF-α在促炎细胞因子释放中的作用。目前的概念依赖于 TNF-α 刺激其他促炎细胞因子的级联释放,例如 IL-1β、IL-6 和 IL-8(大鼠中的 CINC-1),从而触发最终释放炎症介质前列腺素 E 2 (PGE 2) 和直接使伤害感受器敏感的交感神经胺。然而,这可能不是唯一的机制,因为沙利度胺阻断 TNF-α 合成可防止痛觉过敏,而不会中断 IL-1β、IL-6 和 CINC-1 的合成。因此,我们假设 TNF-α 激活 TNF-α 1 型受体 (TNFR1) 会增加伤害感受器对 PGE 2和多巴胺作用的敏感性。我们发现鞘内注射抗 TNFR1 或沙利度胺的寡脱氧核苷酸反义 (ODN-AS) 可预防角叉菜胶诱导的痛觉过敏。TNF-α 与阈下剂量 PGE 2的共同给药在用地塞米松(以防止细胞因子的内源性释放)预处理的 Wistar 大鼠的后爪中,本身不诱导痛觉过敏的多巴胺诱导强烈的痛觉过敏,而用针对 TNFR1 的 ODN-AS 鞘内治疗可以防止这种强烈的痛觉过敏。我们认为 TNF-α 对神经元 TNFR1 的激活决定性地增加了外周传入神经元对最终诱导痛觉过敏的最终炎症介质(PGE 2和多巴胺)作用的敏感性。这种机制也可能是沙利度胺镇痛作用的基础。
更新日期:2020-09-26
中文翻译:
外周组织的炎症性疼痛取决于初级传入神经元中 TNF-α 1 型受体的激活
肿瘤坏死因子α(TNF-α)在炎症性痛觉过敏发展中的作用机制已被广泛研究,主要是TNF-α在促炎细胞因子释放中的作用。目前的概念依赖于 TNF-α 刺激其他促炎细胞因子的级联释放,例如 IL-1β、IL-6 和 IL-8(大鼠中的 CINC-1),从而触发最终释放炎症介质前列腺素 E 2 (PGE 2) 和直接使伤害感受器敏感的交感神经胺。然而,这可能不是唯一的机制,因为沙利度胺阻断 TNF-α 合成可防止痛觉过敏,而不会中断 IL-1β、IL-6 和 CINC-1 的合成。因此,我们假设 TNF-α 激活 TNF-α 1 型受体 (TNFR1) 会增加伤害感受器对 PGE 2和多巴胺作用的敏感性。我们发现鞘内注射抗 TNFR1 或沙利度胺的寡脱氧核苷酸反义 (ODN-AS) 可预防角叉菜胶诱导的痛觉过敏。TNF-α 与阈下剂量 PGE 2的共同给药在用地塞米松(以防止细胞因子的内源性释放)预处理的 Wistar 大鼠的后爪中,本身不诱导痛觉过敏的多巴胺诱导强烈的痛觉过敏,而用针对 TNFR1 的 ODN-AS 鞘内治疗可以防止这种强烈的痛觉过敏。我们认为 TNF-α 对神经元 TNFR1 的激活决定性地增加了外周传入神经元对最终诱导痛觉过敏的最终炎症介质(PGE 2和多巴胺)作用的敏感性。这种机制也可能是沙利度胺镇痛作用的基础。

























 京公网安备 11010802027423号
京公网安备 11010802027423号