Current Bioinformatics ( IF 4 ) Pub Date : 2020-05-31 , DOI: 10.2174/1574893615666200106115421 Peng Chen 1 , Tong Shen 1 , Youzhi Zhang 2 , Bing Wang 3
|
Background: Hotspots are those residues that contribute major free energy of binding in protein-protein interactions. Protein functions are frequently dependent on hotspot residues. At present, hotspot residues are always identified by Alanine scanning mutagenesis technology, which is costly, time-consuming and laborious.
Objective: Therefore, more accurate and efficient methods have to be developed to identify protein hotspot residues.
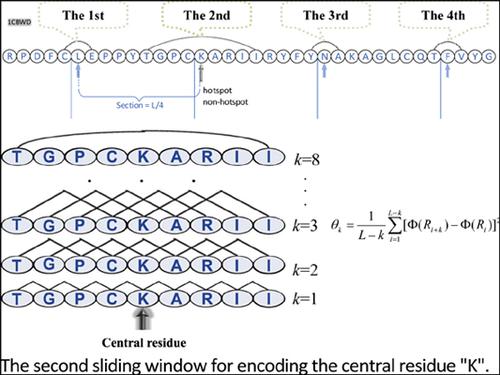
Methods: This paper proposed a novel encoding schema of sequence-segment neighbors and constructed a random forest-based model to identify hotspots in protein interaction interfaces. Firstly, 10 amino acid physicochemical properties, 16 features related to the PI and DI, and 25 features related to ASA were extracted. Different from the previous residue encoding schemas, such as auto correlation descriptor or triplet combination information, this paper employed the influence of amino acids neighbors to hotspot residues and amino acids with a certain distance in sequence to the hotspot.
Results: Moreover, the proposed model was compared with other hotspot prediction methods, including APIS, Robetta, FOLDEF, KFC, MINERVA models, etc.
Conclusion: The experimental results showed that the proposed model can improve the prediction ability of protein hotspot residues on the same test set.
中文翻译:
蛋白质热点残基预测的序列段邻居编码方案。
背景:热点是那些在蛋白质-蛋白质相互作用中贡献主要结合自由能的残基。蛋白质功能通常取决于热点残基。目前,总是通过丙氨酸扫描诱变技术来鉴定热点残留物,该技术昂贵,费时且费力。
目的:因此,必须开发出更准确和有效的方法来鉴定蛋白质热点残基。
方法:本文提出了一种新的序列段邻居编码方案,并构建了一个基于森林的随机模型来识别蛋白质相互作用界面中的热点。首先,提取了10种氨基酸的理化特性,与PI和DI有关的16个特征以及与ASA有关的25个特征。与以前的残基编码模式(例如自相关描述符或三联体组合信息)不同,本文采用了氨基酸邻近区域对热点残基的影响以及氨基酸序列与热点之间有一定距离的影响。
结果:此外,将提出的模型与其他热点预测方法进行了比较,包括APIS,Robetta,FOLDEF,KFC,MINERVA模型等。
结论:实验结果表明,该模型可以提高同一测试集上蛋白质热点残基的预测能力。

























 京公网安备 11010802027423号
京公网安备 11010802027423号