Current Bioinformatics ( IF 4 ) Pub Date : 2020-11-30 , DOI: 10.2174/1574893615666200305090055 Xiaogeng Wan 1 , Xinying Tan 2
|
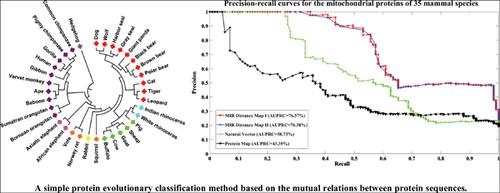
Background: Protein is a kind of important organics in life. It is varied with its sequences, structures and functions. Protein evolutionary classification is one of the popular research topics in computational bioinformatics. Many studies have used protein sequence information to classify the evolutionary relationships of proteins. As the amount of protein sequence data increases, efficient computational tools are needed to make efficient protein evolutionary classifications with high accuracies in the big data paradigm.
Methods: In this study, we propose a new simple and efficient computational approach based on the normalized mutual information rates to compute the relationship between protein sequences, we then use the “distances” defined on the relationships to perform the evolutionary classifications of proteins. The new method is computational efficient, model-free and unsupervised, which does not require training data when performing classifications.
Results: Simulation studies on various examples demonstrate the efficiency of the new method. We use precision-recall curves to compare the efficiency of our new method with traditional methods, results show that the new method outperforms the traditional methods in most of the cases when performing evolutionary classifications.
Conclusion: The new method is simple and proved to be efficient in protein evolutionary classifications, which is useful in future evolutionary analysis particularly in the big data paradigm.
中文翻译:
基于蛋白质序列之间相互关系的简单蛋白质进化分类方法
背景:蛋白质是生命中一种重要的有机物。它的顺序,结构和功能各不相同。蛋白质进化分类是计算生物信息学中流行的研究主题之一。许多研究已经使用蛋白质序列信息对蛋白质的进化关系进行分类。随着蛋白质序列数据量的增加,需要有效的计算工具来在大数据范式中进行具有高精度的高效蛋白质进化分类。
方法:在这项研究中,我们提出了一种基于归一化互信息率的新的简单有效的计算方法来计算蛋白质序列之间的关系,然后我们使用在这些关系上定义的“距离”来进行蛋白质的进化分类。新方法计算效率高,无模型且不受监督,在执行分类时不需要训练数据。
结果:对各种示例的仿真研究证明了该新方法的有效性。我们使用精确召回曲线将新方法与传统方法的效率进行比较,结果表明,在进行进化分类的大多数情况下,新方法的性能优于传统方法。
结论:该新方法简单易行,被证明在蛋白质进化分类中是有效的,可用于未来的进化分析,特别是在大数据范例中。
























 京公网安备 11010802027423号
京公网安备 11010802027423号