Journal of Nanoparticle Research ( IF 2.5 ) Pub Date : 2020-09-22 , DOI: 10.1007/s11051-020-05021-3 Olubunmi Kolawole Akiode , Palanichamy Murugan , Abideen Idowu Adeogun , Gboyega Augustine Adebayo , Mopelola Abidemi Idowu
|
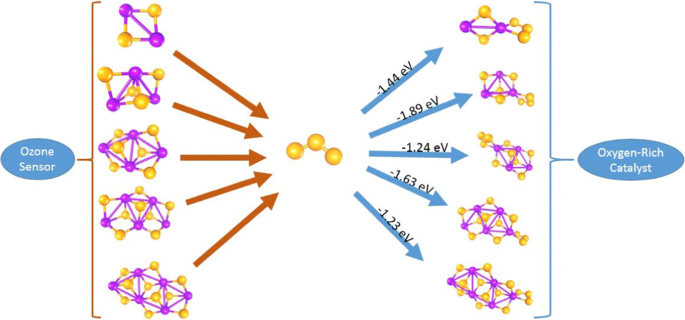
A systematic conceptual study of the properties of tin oxide nanoclusters SnnOm (n = 1–6; 2n+1 ≤ m ≤ 4n+2) with ozonide ion or mixed ozonide with molecular O20, oxide (O2−), peroxide (O−) or superoxide (O2−) was investigated using the ab initio density functional theory perspective. Bader charge calculations revealed the oxidation state of Sn and established formation of terminal oxide moiety in the nanoclusters. The stability investigation was carried out by calculations of the binding energy (BE) per atom, the second difference total energy (Δ2E(m)) and the gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) while adsorption energy per O2 and O3 was investigated. Exploring the variation of the reactivity parameters and reaction energy with sizes revealed size dependence on physicochemical properties of the nanoclusters. Magnetism studies show the contribution of O2, O−, O2− and O3− to the magnetic moment of the nanoclusters.
Graphical abstract
中文翻译:
锡氧化物纳米簇的结构演变,电子和物理化学性质:密度泛函理论的观点
的氧化锡Sn的纳米团簇的性质的系统的概念研究Ñ ø米(Ñ = 1-6; 2 Ñ 1≤米≤4 Ñ 2)用的臭氧化物离子或混合的臭氧化物用分子直径:2 0,氧化物(O 2 - ),过氧化(O - ),或过氧化物(O 2 - ),使用从头密度泛函理论透视进行了研究。较差的电荷计算揭示了Sn的氧化态并在纳米簇中确定了末端氧化物部分的形成。通过计算每个原子的结合能(BE),第二差总能量(Δ研究了2 E(m))和最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)之间的间隙,同时研究了每O 2和O 3的吸附能。探索反应性参数和反应能量随尺寸的变化,发现尺寸取决于纳米团簇的物理化学性质。磁性研究表演O的贡献2,O- -,O- 2 -和O 3 -对磁性纳米团簇的时刻。
图形概要

























 京公网安备 11010802027423号
京公网安备 11010802027423号