Interdisciplinary Sciences: Computational Life Sciences ( IF 4.8 ) Pub Date : 2020-09-21 , DOI: 10.1007/s12539-020-00387-3 Tianyang Yu 1, 2 , Na Xu 1, 2 , Neshatul Haque 3 , Chang Gao 1 , Wenhua Huang 1, 4 , Zunnan Huang 1, 2, 5
|
Abstract
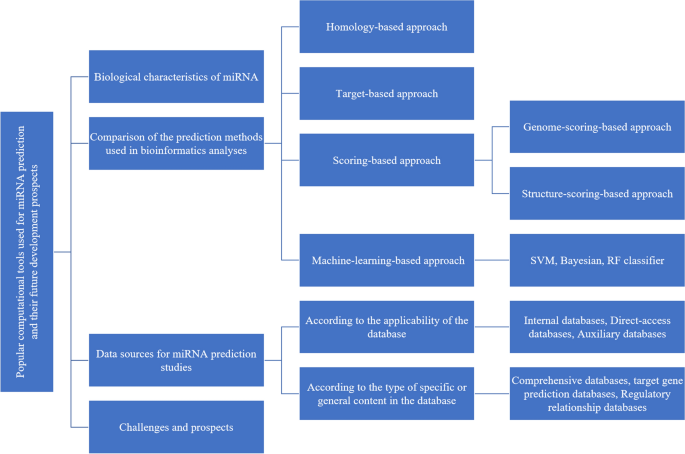
MicroRNAs (miRNAs) are 19–24 nucleotide (nt)-long noncoding, single-stranded RNA molecules that play significant roles in regulating the gene expression, growth, and development of plants and animals. From the year that miRNAs were first discovered until the beginning of the twenty-first century, researchers used experimental methods such as cloning and sequencing to identify new miRNAs and their roles in the posttranscriptional regulation of protein synthesis. Later, in the early 2000s, informatics approaches to the discovery of new miRNAs began to be implemented. With increasing knowledge about miRNA, more efficient algorithms have been developed for computational miRNA prediction. The miRNA research community, hoping for greater coverage and faster results, has shifted from cumbersome and expensive traditional experimental approaches to computational approaches. These computational methods started with homology-based comparisons of known miRNAs with orthologs in the genomes of other species; this method could identify a known miRNA in new species. Second-generation sequencing and next-generation sequencing of mRNA at different developmental stages and in specific tissues, in combination with a better search and alignment algorithm, have accelerated the process of predicting novel miRNAs in a particular species. Using the accumulated annotated miRNA sequence information, researchers have been able to design ab initio algorithms for miRNA prediction independent of genome sequence knowledge. Here, the methods recently used for miRNA computational prediction are summarized and classified into the following four categories: homology-based, target-based, scoring-based, and machine-learning-based approaches. Finally, the future developmental directions of miRNA prediction methods are discussed.
Graphic Abstract
中文翻译:
用于 miRNA 预测的流行计算工具及其未来发展前景。
摘要
MicroRNA (miRNA) 是 19-24 个核苷酸 (nt) 长的非编码单链 RNA 分子,在调节植物和动物的基因表达、生长和发育方面发挥着重要作用。从 miRNA 首次被发现到 21 世纪初,研究人员使用克隆和测序等实验方法来鉴定新的 miRNA 及其在蛋白质合成的转录后调控中的作用。后来,在 2000 年代初,开始实施发现新 miRNA 的信息学方法。随着对 miRNA 的了解越来越多,已经开发了更有效的算法来计算 miRNA 预测。miRNA 研究界,希望更大的覆盖范围和更快的结果,已经从繁琐且昂贵的传统实验方法转变为计算方法。这些计算方法始于将已知 miRNA 与其他物种基因组中的直向同源物进行基于同源性的比较;这种方法可以识别新物种中的已知miRNA。在不同发育阶段和特定组织中对 mRNA 进行二代测序和二代测序,结合更好的搜索和比对算法,加速了预测特定物种中新 miRNA 的过程。使用累积的带注释的 miRNA 序列信息,研究人员已经能够设计出独立于基因组序列知识的 miRNA 预测算法。这里,最近用于 miRNA 计算预测的方法被总结并分为以下四类:基于同源性的方法、基于目标的方法、基于评分的方法和基于机器学习的方法。最后,讨论了miRNA预测方法的未来发展方向。


























 京公网安备 11010802027423号
京公网安备 11010802027423号