当前位置:
X-MOL 学术
›
Clin. Exp. Immunol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Clinical, functional and genetic characterization of Sixteen Patients Suffering from Chronic Granulomatous Disease variants - Identification of Eleven Novel Mutations in CYBB.
Clinical & Experimental Immunology ( IF 4.6 ) Pub Date : 2020-09-20 , DOI: 10.1111/cei.13520 M Mollin 1 , S Beaumel 1 , B Vigne 1 , J Brault 1 , N Roux-Buisson 2, 3 , J Rendu 2, 3 , V Barlogis 4 , G Catho 5 , C Dumeril 6 , F Fouyssac 7 , D Monnier 8 , V Gandemer 9 , M Revest 10 , J-P Brion 11 , C Bost-Bru 12 , E Jeziorski 13 , L Eitenschenck 6 , C Jarrasse 6 , S Drillon Haus 14 , M Houachée-Chardin 5 , M Hancart 13 , G Michel 4 , Y Bertrand 5 , D Plantaz 12 , J Kelecic 15 , R Traberg 16 , L Kainulainen 17, 18 , J Fauré 2, 3 , F Fieschi 19 , M J Stasia 1, 19
Clinical & Experimental Immunology ( IF 4.6 ) Pub Date : 2020-09-20 , DOI: 10.1111/cei.13520 M Mollin 1 , S Beaumel 1 , B Vigne 1 , J Brault 1 , N Roux-Buisson 2, 3 , J Rendu 2, 3 , V Barlogis 4 , G Catho 5 , C Dumeril 6 , F Fouyssac 7 , D Monnier 8 , V Gandemer 9 , M Revest 10 , J-P Brion 11 , C Bost-Bru 12 , E Jeziorski 13 , L Eitenschenck 6 , C Jarrasse 6 , S Drillon Haus 14 , M Houachée-Chardin 5 , M Hancart 13 , G Michel 4 , Y Bertrand 5 , D Plantaz 12 , J Kelecic 15 , R Traberg 16 , L Kainulainen 17, 18 , J Fauré 2, 3 , F Fieschi 19 , M J Stasia 1, 19
Affiliation
|
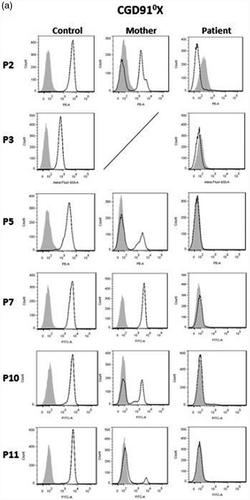
Chronic granulomatous disease (CGD) is a rare inherited disorder in which phagocytes lack nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. The most common form is the X‐linked CGD (X91‐CGD), caused by mutations in the CYBB gene. Clinical, functional and genetic characterizations of 16 CGD cases of male patients and their relatives were performed. We classified them as suffering from different variants of CGD (X910, X91− or X91+), according to NADPH oxidase 2 (NOX2) expression and NADPH oxidase activity in neutrophils. Eleven mutations were novel (nine X910‐CGD and two X91−‐CGD). One X910‐CGD was due to a new and extremely rare double missense mutation Thr208Arg‐Thr503Ile. We investigated the pathological impact of each single mutation using stable transfection of each mutated cDNA in the NOX2 knock‐out PLB‐985 cell line. Both mutations leading to X91−‐CGD were also novel; one deletion, c.‐67delT, was localized in the promoter region of CYBB; the second c.253‐1879A>G mutation activates a splicing donor site, which unveils a cryptic acceptor site leading to the inclusion of a 124‐nucleotide pseudo‐exon between exons 3 and 4 and responsible for the partial loss of NOX2 expression. Both X91−‐CGD mutations were characterized by a low cytochrome b558 expression and a faint NADPH oxidase activity. The functional impact of new missense mutations is discussed in the context of a new three‐dimensional model of the dehydrogenase domain of NOX2. Our study demonstrates that low NADPH oxidase activity found in both X91−‐CGD patients correlates with mild clinical forms of CGD, whereas X910‐CGD and X91+‐CGD cases remain the most clinically severe forms.
中文翻译:

16 名患有慢性肉芽肿病变异的患者的临床、功能和遗传特征 - CYBB 中 11 种新突变的鉴定。
慢性肉芽肿病 (CGD) 是一种罕见的遗传性疾病,其中吞噬细胞缺乏烟酰胺腺嘌呤二核苷酸磷酸 (NADPH) 氧化酶活性。最常见的形式是由CYBB基因突变引起的 X 连锁 CGD (X91-CGD) 。对 16 例男性患者及其亲属的 CGD 病例进行临床、功能和遗传特征分析。根据中性粒细胞中 NADPH 氧化酶 2 (NOX2) 的表达和 NADPH 氧化酶活性,我们将它们分类为患有不同的 CGD 变体(X91 0、X91 -或 X91 + )。11 个突变是新的(9 个 X91 0 -CGD 和两个 X91 - -CGD)。一个 X91 0‐CGD 是由于一种新的极其罕见的双错义突变 Thr208Arg‐Thr503Ile。我们使用 NOX2 敲除 PLB-985 细胞系中每个突变 cDNA 的稳定转染研究了每个单个突变的病理影响。导致 X91 - -CGD 的两种突变也是新的。一个缺失,c.-67delT,位于 CYBB 的启动子区域;第二个 c.253-1879A>G 突变激活了一个剪接供体位点,该位点揭示了一个神秘的受体位点,导致在外显子 3 和 4 之间包含一个 124 个核苷酸的假外显子,并导致 NOX2 表达的部分丧失。两种 X91 - -CGD 突变均以低细胞色素b 558为特征表达和微弱的NADPH氧化酶活性。在新的 NOX2 脱氢酶结构域三维模型的背景下讨论了新错义突变的功能影响。我们的研究表明,在 X91 - -CGD 患者中发现的低 NADPH 氧化酶活性与 CGD 的轻度临床形式相关,而 X91 0 -CGD 和 X91 + -CGD 病例仍然是临床上最严重的形式。
更新日期:2020-09-20
中文翻译:
16 名患有慢性肉芽肿病变异的患者的临床、功能和遗传特征 - CYBB 中 11 种新突变的鉴定。
慢性肉芽肿病 (CGD) 是一种罕见的遗传性疾病,其中吞噬细胞缺乏烟酰胺腺嘌呤二核苷酸磷酸 (NADPH) 氧化酶活性。最常见的形式是由CYBB基因突变引起的 X 连锁 CGD (X91-CGD) 。对 16 例男性患者及其亲属的 CGD 病例进行临床、功能和遗传特征分析。根据中性粒细胞中 NADPH 氧化酶 2 (NOX2) 的表达和 NADPH 氧化酶活性,我们将它们分类为患有不同的 CGD 变体(X91 0、X91 -或 X91 + )。11 个突变是新的(9 个 X91 0 -CGD 和两个 X91 - -CGD)。一个 X91 0‐CGD 是由于一种新的极其罕见的双错义突变 Thr208Arg‐Thr503Ile。我们使用 NOX2 敲除 PLB-985 细胞系中每个突变 cDNA 的稳定转染研究了每个单个突变的病理影响。导致 X91 - -CGD 的两种突变也是新的。一个缺失,c.-67delT,位于 CYBB 的启动子区域;第二个 c.253-1879A>G 突变激活了一个剪接供体位点,该位点揭示了一个神秘的受体位点,导致在外显子 3 和 4 之间包含一个 124 个核苷酸的假外显子,并导致 NOX2 表达的部分丧失。两种 X91 - -CGD 突变均以低细胞色素b 558为特征表达和微弱的NADPH氧化酶活性。在新的 NOX2 脱氢酶结构域三维模型的背景下讨论了新错义突变的功能影响。我们的研究表明,在 X91 - -CGD 患者中发现的低 NADPH 氧化酶活性与 CGD 的轻度临床形式相关,而 X91 0 -CGD 和 X91 + -CGD 病例仍然是临床上最严重的形式。


























 京公网安备 11010802027423号
京公网安备 11010802027423号