当前位置:
X-MOL 学术
›
Polym. Int.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Sorption-induced polymer rearrangement: approaches from molecular modeling
Polymer International ( IF 3.2 ) Pub Date : 2020-09-13 , DOI: 10.1002/pi.6124 Dylan M Anstine 1, 2 , Coray M Colina 1, 2, 3
Polymer International ( IF 3.2 ) Pub Date : 2020-09-13 , DOI: 10.1002/pi.6124 Dylan M Anstine 1, 2 , Coray M Colina 1, 2, 3
Affiliation
|
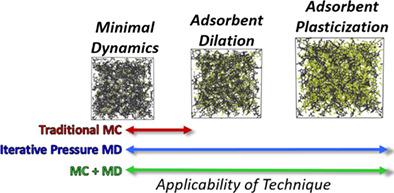
With the growing need for chemical separation and chemical storage solutions, polymeric adsorbents have emerged as a promising class of candidate materials because of their potentially tunable sorption properties, membrane structure and relatively cost consciousness. Moreover, the developing field of polymeric membrane materials has shown particular success at integrating both experimental and computational studies. However, these material systems are known to suffer from varying degrees of induced membrane structural rearrangement upon adsorbate uptake, and thus many polymeric membrane performance metrics are often considered to degrade with an increasing number of ‘guest’ species. In this mini-review, we highlight methodology tradeoffs and provide insights into atomistic molecular simulations used to study adsorption with flexible frameworks, which have the potential to predict separation, storage or catalytic capabilities a priori to experimental efforts. Specifically, molecular simulation methods that have been applied to provide predictions of polymeric membrane properties that have included consideration for sorbate-induced polymer chain rearrangement, swelling and/or plasticization are reviewed. The examples and methodologies described provide demonstrations of the applicability of simulations as an approach to understand adsorption-based phenomena at an atomistic/molecular level, and as a tool to carry out screening studies aimed at efficiently providing analysis for a diversity of polymeric adsorbent–adsorbate systems. © 2020 Society of Industrial Chemistry
中文翻译:

吸附诱导的聚合物重排:来自分子建模的方法
随着对化学分离和化学储存解决方案的需求不断增长,聚合物吸附剂因其潜在的可调节吸附性能、膜结构和相对成本意识而成为一类有前途的候选材料。此外,聚合物膜材料的发展领域在整合实验和计算研究方面表现出特别的成功。然而,已知这些材料系统在吸附质吸收时会遭受不同程度的诱导膜结构重排,因此许多聚合物膜性能指标通常被认为会随着“客体”物种数量的增加而降低。在这篇小型评论中,我们强调了方法论的权衡,并提供了对用于研究具有灵活框架的吸附的原子分子模拟的见解,实验工作的先验。具体而言,回顾了已应用于提供聚合物膜性能预测的分子模拟方法,其中包括考虑山梨酸酯诱导的聚合物链重排、溶胀和/或增塑。所描述的示例和方法证明了模拟作为一种在原子/分子水平上理解基于吸附的现象的方法的适用性,以及作为进行筛选研究的工具,旨在有效地分析各种聚合物吸附剂 - 被吸附物系统。© 2020 工业化学学会
更新日期:2020-09-13
中文翻译:
吸附诱导的聚合物重排:来自分子建模的方法
随着对化学分离和化学储存解决方案的需求不断增长,聚合物吸附剂因其潜在的可调节吸附性能、膜结构和相对成本意识而成为一类有前途的候选材料。此外,聚合物膜材料的发展领域在整合实验和计算研究方面表现出特别的成功。然而,已知这些材料系统在吸附质吸收时会遭受不同程度的诱导膜结构重排,因此许多聚合物膜性能指标通常被认为会随着“客体”物种数量的增加而降低。在这篇小型评论中,我们强调了方法论的权衡,并提供了对用于研究具有灵活框架的吸附的原子分子模拟的见解,实验工作的先验。具体而言,回顾了已应用于提供聚合物膜性能预测的分子模拟方法,其中包括考虑山梨酸酯诱导的聚合物链重排、溶胀和/或增塑。所描述的示例和方法证明了模拟作为一种在原子/分子水平上理解基于吸附的现象的方法的适用性,以及作为进行筛选研究的工具,旨在有效地分析各种聚合物吸附剂 - 被吸附物系统。© 2020 工业化学学会


























 京公网安备 11010802027423号
京公网安备 11010802027423号