当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Utilizing Co-Crystallization as a Tool to Unravel the Structural Diversity and Electronic Features of I···S Halogen Bonded Interactions in Stoichiomorphic Co-Crystals
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2020-09-10 , DOI: 10.1021/acs.cgd.0c00823 Avantika Hasija 1 , Deepak Chopra 1
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2020-09-10 , DOI: 10.1021/acs.cgd.0c00823 Avantika Hasija 1 , Deepak Chopra 1
Affiliation
|
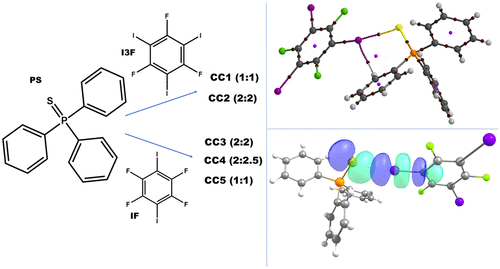
In the current investigation, we report five new co-crystals formed by combinations of triphenylphosphine sulfide (PS) with 1,3,5-triiodo-2,4,6-trifluorobenzene (I3F), triphenylphosphine sulfide (PS) with 1,4-diiodotetrafluorobenzene (IF) which consists of I-centered interactions, primarily consisting of I···S halogen bonding interactions. The distance between the iodine and sulfur atoms in the different crystal structures is much shorter than the sum of the van der Waals radii (shortest I···S distance, 3.163 Å) and is highly directional (most directional C–I···S, 179°) in nature. The three-dimensional deformation density shows that the charge depleted (CD) region on iodine electrostatically interacts with the charge concentrated (CC) region on sulfur. The interaction energy, as obtained from density functional theory calculations, is in the range of −16 to −32 kJ/mol, for the interacting units, at the crystal geometry. The atomic polarizability analysis establishes the mutual polarization of the iodine atom in the presence of the sulfur atom and vice versa. A topological analysis unequivocally establishes the presence of a (3, −1) bond critical point, and the NCI-RDG analysis establishes the attractive nature of the interaction. The elongation of the C–I bond length as a consequence of evident charge transfer from sulfur to iodine for I···S interaction is computed via natural bonding orbitals. Thus, a detailed computational analysis renders insights into the electronic nature of the observed I···S interactions in the solid state.
中文翻译:

利用共结晶作为工具来解理纯同晶共晶体中I··S卤素键相互作用的结构多样性和电子特征
在当前的调查中,我们报告了由三苯基膦硫化物(PS)与1,3,5-三碘代-2,4,6-三氟苯(I3F),三苯基膦硫化物(PS)与1,4结合形成的五个新的共晶体-二碘四氟苯(IF),它由以I为中心的相互作用组成,主要由I··S卤素键相互作用组成。碘和硫原子在不同晶体结构中的距离比范德华半径之和(最短的I··S距离,3.163Å)短得多,并且具有高度的方向性(最方向的C–I···· S,179°)。三维变形密度表明,碘上的电荷耗尽(CD)区与硫上的电荷集中(CC)区发生静电相互作用。从密度泛函理论计算中获得的相互作用能,对于相互作用的单元,在晶体几何形状下,其在-16至-32kJ / mol的范围内。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I···S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。NCI-RDG分析建立了互动的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。NCI-RDG分析建立了互动的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。
更新日期:2020-10-07
中文翻译:
利用共结晶作为工具来解理纯同晶共晶体中I··S卤素键相互作用的结构多样性和电子特征
在当前的调查中,我们报告了由三苯基膦硫化物(PS)与1,3,5-三碘代-2,4,6-三氟苯(I3F),三苯基膦硫化物(PS)与1,4结合形成的五个新的共晶体-二碘四氟苯(IF),它由以I为中心的相互作用组成,主要由I··S卤素键相互作用组成。碘和硫原子在不同晶体结构中的距离比范德华半径之和(最短的I··S距离,3.163Å)短得多,并且具有高度的方向性(最方向的C–I···· S,179°)。三维变形密度表明,碘上的电荷耗尽(CD)区与硫上的电荷集中(CC)区发生静电相互作用。从密度泛函理论计算中获得的相互作用能,对于相互作用的单元,在晶体几何形状下,其在-16至-32kJ / mol的范围内。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。原子极化率分析建立了在硫原子存在下碘原子的相互极化,反之亦然。拓扑分析明确地确定了(3,-1)键临界点的存在,而NCI-RDG分析则确定了相互作用的吸引力。由于I···S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。NCI-RDG分析建立了互动的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。NCI-RDG分析建立了互动的吸引力。由于I··S相互作用而从硫到碘的明显电荷转移,C–I键长度的延长是通过自然键合轨道计算的。因此,详细的计算分析使我们深入了解了固态下观察到的I·S相互作用的电子性质。

























 京公网安备 11010802027423号
京公网安备 11010802027423号