当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Nonadiabatic dynamics in multidimensional complex potential energy surfaces
Chemical Science ( IF 8.4 ) Pub Date : 2020-09-07 , DOI: 10.1039/d0sc04197a Fábris Kossoski 1, 2, 3, 4 , Mario Barbatti 1, 2, 3, 4
Chemical Science ( IF 8.4 ) Pub Date : 2020-09-07 , DOI: 10.1039/d0sc04197a Fábris Kossoski 1, 2, 3, 4 , Mario Barbatti 1, 2, 3, 4
Affiliation
|
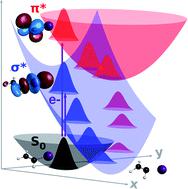
Despite the continuous development of theoretical methodologies for describing nonadiabatic dynamics of molecular systems, there is a lack of approaches for processes where the norm of the wave function is not conserved, i.e., when an imaginary potential accounts for some irreversible decaying mechanism. Current approaches rely on building potential energy surfaces of reduced dimensionality, which is not optimal for more involving and realistic multidimensional problems. Here, we present a novel methodology for describing the dynamics of complex-valued molecular Hamiltonians, which is a generalisation of the trajectory surface hopping method. As a first application, the complex surface fewest switches surface hopping (CS-FSSH) method was employed to survey the relaxation mechanisms of the shape resonant anions of iodoethene. We have provided the first detailed and dynamical picture of the π*/σ* mechanism of dissociative electron attachment in halogenated unsaturated compounds, which is believed to underlie electron-induced reactions of several molecules of interest. Electron capture into the π* orbital promotes C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and out-of-plane vibrations, followed by charge transfer from the double bond into the σ* orbital at the C–I bond, and, finally, release of the iodine ion, all within only 15 fs. On-the-fly dynamics simulations of a vast class of processes can be envisioned with the CS-FSSH methodology, including autoionisation from transient anions, core-ionised and superexcited states, Auger and interatomic coulombic decay, and time-dependent luminescence.
C stretching and out-of-plane vibrations, followed by charge transfer from the double bond into the σ* orbital at the C–I bond, and, finally, release of the iodine ion, all within only 15 fs. On-the-fly dynamics simulations of a vast class of processes can be envisioned with the CS-FSSH methodology, including autoionisation from transient anions, core-ionised and superexcited states, Auger and interatomic coulombic decay, and time-dependent luminescence.
中文翻译:

多维复杂势能面的非绝热动力学
尽管用于说明分子系统的非绝热动力学理论方法的不断发展,存在缺乏对过程的方法,其中所述波函数的范数不守恒,即,当假想势解释某些不可逆的衰减机制时。当前的方法依赖于构建尺寸减小的势能表面,这对于更多涉及和现实的多维问题不是最佳的。在这里,我们提出了一种新颖的方法来描述复杂值分子哈密顿量的动力学,这是对轨迹表面跳变方法的概括。作为首次应用,采用复杂表面最少开关表面跳变(CS-FSSH)方法来研究碘乙烯的形状共振阴离子的弛豫机理。我们提供了卤代不饱和化合物中电子离解的π* /σ*机理的第一张详细的动力学图,据信这是感兴趣的几个分子的电子诱导反应的基础。C拉伸和平面外振动,然后电荷从双键转移到C–I键的σ*轨道,最后释放碘离子,所有这些仅在15 fs内完成。可以使用CS-FSSH方法对大量过程进行动态动力学模拟,包括从瞬态阴离子,核心电离态和超激发态,俄歇和原子间库仑衰变以及随时间变化的发光进行自动电离。
更新日期:2020-09-23
C stretching and out-of-plane vibrations, followed by charge transfer from the double bond into the σ* orbital at the C–I bond, and, finally, release of the iodine ion, all within only 15 fs. On-the-fly dynamics simulations of a vast class of processes can be envisioned with the CS-FSSH methodology, including autoionisation from transient anions, core-ionised and superexcited states, Auger and interatomic coulombic decay, and time-dependent luminescence.
中文翻译:
多维复杂势能面的非绝热动力学
尽管用于说明分子系统的非绝热动力学理论方法的不断发展,存在缺乏对过程的方法,其中所述波函数的范数不守恒,即,当假想势解释某些不可逆的衰减机制时。当前的方法依赖于构建尺寸减小的势能表面,这对于更多涉及和现实的多维问题不是最佳的。在这里,我们提出了一种新颖的方法来描述复杂值分子哈密顿量的动力学,这是对轨迹表面跳变方法的概括。作为首次应用,采用复杂表面最少开关表面跳变(CS-FSSH)方法来研究碘乙烯的形状共振阴离子的弛豫机理。我们提供了卤代不饱和化合物中电子离解的π* /σ*机理的第一张详细的动力学图,据信这是感兴趣的几个分子的电子诱导反应的基础。
C拉伸和平面外振动,然后电荷从双键转移到C–I键的σ*轨道,最后释放碘离子,所有这些仅在15 fs内完成。可以使用CS-FSSH方法对大量过程进行动态动力学模拟,包括从瞬态阴离子,核心电离态和超激发态,俄歇和原子间库仑衰变以及随时间变化的发光进行自动电离。

























 京公网安备 11010802027423号
京公网安备 11010802027423号