Immunology and Cell Biology ( IF 4 ) Pub Date : 2020-09-07 , DOI: 10.1111/imcb.12388 Adrian Hoffmann 1, 2 , Jürgen Bernhagen 1, 3, 4
|
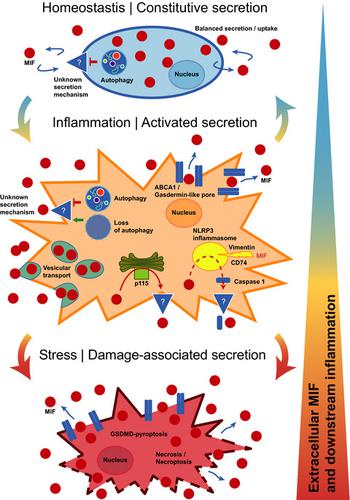
Cytokines are protein mediators regulating intercellular communication. They orchestrate the immune response and play pivotal roles in numerous diseases such as inflammatory and autoimmune diseases, cardiovascular conditions and cancer. Understanding their expression and secretion mechanisms therefore is important and conditional to the identification of therapeutic strategies blocking the release of pathogenic cytokines. Most cytokines carry an N‐terminal signal peptide and are secreted via the conventional endoplasmic reticulum (ER)‐Golgi route. However, a number of important inflammatory mediators including the interleukin (IL)‐1 family of cytokines do not express a signal peptide and are released by alternative pathways, collectively termed unconventional protein secretion (UPS). Macrophage migration‐inhibitory factor (MIF) is one of the oldest cytokines discovered. It is a key upstream regulator of innate immunity and an important host defense protein, whereas dysregulated MIF is a pivotal mediator of acute and chronic inflammatory conditions, atherosclerosis, rheumatoid arthritis and systemic lupus erythematosus (SLE).1-3 MIF is an “atypical” cytokine; this notion refers not only to its high evolutionary conservation across kingdoms, but also to molecular characteristics such as an N‐terminal tautomerase pocket, its chemokine‐like behavior and engagement of CXC chemokine receptors, lack of a signal peptide and abundant cytosolic expression as well as presumed additional intracellular functions.4 In fact, MIF was suggested to be released by unconventional protein secretion routes previously,5 but ultimate evidence has been missing and the precise mechanisms have remained incompletely understood. This study by Dankers et al. adds an important new facet to its secretion mechanism, linking MIF release to the processes of necroptosis and nucleotide‐binding domain, leucine‐rich‐repeat‐containing (NLR) family pyrin domain containing 3 (NLRP3) inflammasome‐dependent pyroptosis.6 The data contribute to establish MIF as a member of the “unconventional protein secretion UPS club of proteins.”
MIF was discovered as a T‐cell and macrophage cytokine, but it is now clear that it is broadly expressed in most cell types. Abundant cytosolic storage pools of MIF and baseline (nonpathologic) circulating MIF levels in the 1–10 ng mL−1 range are considered to be part of its atypical cytokine profile. Under inflammatory conditions, circulating MIF levels are rapidly and drastically increased, reaching levels up to 500–1500 ng mL−1. The actual local tissue levels of released MIF protein under disease conditions are poorly defined, but in vitro cell culture‐based studies suggest a similar rate of elevation by up to several 10‐ to 100‐fold. While MIF secretion studies have mainly been performed in immune cells, it has become clear that secretion can also occur in response to a variety of stimuli from endothelial cells and platelets, parenchymal cells such as cardiomyocytes, vascular smooth muscle cells and fibroblasts as well as tumor cells. That the mechanism(s) underlying the upregulation of MIF release is unconventional and endoplasmic reticulum/Golgi independent was initially insinuated by the lack of a signal peptide in the MIF sequence and was experimentally demonstrated by studies using inhibitors of the classical ER/Golgi pathway.7, 8 Protein interaction studies and the application of pharmacological inhibitors implicated the ABC transporter ABCA1, the signalosome subunit JAB1/CSN5 and the cytosolic, Golgi‐associated protein p115 in the unconventional protein secretion route of MIF.9 JAB1/CSN5 has been suggested to have a sequestration function and p115 facilitates MIF secretion, but it has remained unclear whether these proteins are directly or indirectly involved, and the actual mechanism mediating the release or transport of MIF from the intracellular compartment into the extracellular space has remained undefined.4 Studying immortalized mouse bone marrow‐derived macrophages (BMDM) and human THP‐1 monocyte‐like cells, Dankers et al. now offer evidence that MIF release from monocytes/macrophages may be mediated by necrosis, necroptosis and pyroptosis.6
Necrosis was induced by hydrogen peroxide, ethanol or ultraviolet light, and led to a 3‐ to 100‐fold increase in extracellular MIF over a treatment time of 6 h. Necrosis‐induced release of MIF was shown before for cardiomyocytes and neutrophils10, 11 and actually is expected for an atypical cytokine like MIF that is semiconstitutively expressed and stored intracellularly at relatively high concentrations. In fact, the release pattern following necrotic stimulation of macrophages mirrored that of lactate dehydrogenase (LDH) as a representative of an intracellular house‐keeping protein.6
Necroptosis is a programmed form of necrosis, also considered as regulated inflammatory cell death. Unlike in necrosis, permeabilization of the cell membrane during necroptosis is tightly regulated and is mediated by caspase‐independent receptor‐interacting kinase‐1(RIPK1)/RIPK3/ripoptosome/mixed lineage kinase domain‐like pseudokinase (MLKL)‐mediated pore formation. Dankers et al. treated their bone marrow‐derived macrophages and THP‐1 monocytes with lipopolysaccharide (LPS)/pan‐caspase inhibitor Z‐VAD and TNF inhibitor/inhibitor of apoptosis (IAP) family of proteins BV‐6/Z‐VAD, respectively, to induce necroptosis. They observed a rapid massive release of MIF, which was specifically dependent on the necroptotic signaling machinery as verified by experiments using the RIPK1 inhibitor necrostatin‐1. This is the first report causally linking MIF release from immune cells to necroptosis (Figure 1). Interestingly, a connection between MIF and necroptosis has been suggested before. We previously showed that MIF signaling increases necroptosis in cardiac fibroblasts but protects from necroptosis in tubular epithelial cells in the setting of acute kidney injury.12, 13 Barnes et al. implicated necroptosis in ethanol‐induced receptor‐interacting kinase‐3 RIPK3‐dependent MIF release from liver cells.14 In conjunction with these studies, the current work by Dankers et al. may be suggestive of an intriguing broader role of MIF in necroptotic cell death processes. MIF release is rapidly facilitated by necroptosis, while extracellular MIF in turn controls this programmed inflammatory cell death process.

Pyroptosis is another form of programmed necrotic cell death, shares similarities with necroptosis, but is restricted to immune cells and has a much higher inflammatory potential. Key mechanistic differences are (i) the initiation by the NLRP3 inflammasome (the “pyroptosome”) by intracellular danger signals instead of an extracellular trigger; (ii) the involvement of caspases (caspase‐1/4/5 in humans and caspase‐11 in mice), which lead to the processing and activation of IL‐1 family cytokines and (iii) proteolytic generation of gasdermin D (GSDMD), which is the pore‐forming protein in pyroptotic processes in contrast to MLKLs mixed lineage kinase domain‐like pseudokinases in necroptosis.15 Dankers et al. triggered NLRP3‐mediated pyroptosis in their macrophage cell models by a combination of LPS priming and the potassium ionophore and NLRP3 activator nigericin. They observed marked MIF release under these conditions and verified pyroptosis specificity by the NLRP3‐specific inhibitor MCC950, which abrogated MIF release. While MIF has been previously linked to NLRP3‐associated inflammation (Figure 1), the current paper by Dankers et al. in Immunology & Cell Biology is the first report establishing mechanistic causality between NLRP3 inflammasome activity and MIF secretion. The same authors previously found that MIF functions as an upstream activator of NLRP3, enhancing IL‐1β secretion.16 Surprisingly, this was directly mediated by intracellular MIF, which is required for the interaction between NLRP3 and the intermediate filament protein vimentin. Kang et al. uncovered an alternative mechanism of upstream control of NLRP3 by MIF in human monocytes in the context of lupus autoimmunity. They showed that U1 small‐nucleotide ribonucleoparticle immune complex is a specific trigger of MIF secretion, which subsequently activates NLRP3, caspase‐1 and downstream IL‐1β in a CD74‐dependent manner.17 This upstream control mechanism of NLRP3 thus is a function of autocrine, extracellular MIF (Figure 1). In addition, a potential connection between MIF and the inflammasome had become apparent in a previous iTRAQ mass spectrometry secretome screen, in which MIF was one of the top‐ranked proteins cosecreted with caspase‐1. Although no follow‐up biochemistry was undertaken to confirm that MIF interacts with caspase‐1 and/or that MIF secretion was causally caspase‐1 dependent, the study implicated the NLRP3 inflammasome in MIF secretion18 (Figure 1). Together with the current work by Dankers et al., these studies establish MIF as both an upstream regulator of NLRP3 activity and a novel target of the NLRP3‐dependent pyroptotic cytokine secretion machinery.
This leads over to an intriguing question that neither Dankers et al. nor any of the other studies have yet addressed: how, mechanistically, does intracellular MIF protein that is released during necroptosis or pyroptosis (or through the other suggested pathways; Figure 1) cross the plasma membrane barrier to reach the extracellular compartment? The membrane‐permeabilizing pore that enables for the release of intracellular expressed danger‐associated molecular patterns (DAMPs, also termed alarmis) during necroptosis is MLKL. The release of IL‐1β, other IL‐1 cytokines and DAMPs from inflamed macrophages in pyroptotic cell activation is mediated via membrane pores formed by GSDMD. According to a recent concept, “small” GSDMD pores can act as channels that facilitate the release of IL‐1β and other small leaderless cytokines from viable cells with otherwise intact plasma membranes, whereas “larger” GSDMD pores would be associated with lytic forms of cell content release in more advanced stages of cell hyperactivation.19, 20 The pore molecule executing the release of MIF in activated monocyte/macrophages has yet to be discovered. Figure 1 (middle panel) summarizes the mechanistic options. As Dankers et al. find MIF release to be NLRP3 dependent, GSDMD pores are likely to form concurrent with MIF release, and future studies will need to determine whether GSDMD can function as the MIF‐transporting pore. Alternatively, the previously implicated ABCA1 transporter or an unknown channel/pore may serve as a MIF‐releasing pore. Previous studies with pharmacological inhibitors also have implicated a vesicular export mechanism, the details of which have remained unclear.5 Moreover, some of the experiments performed by Dankers et al. suggest that MIF release may significantly increase in more the advanced, “lytic,” stages of pyroptosis and/or necroptosis. At this stage, membrane translocation of MIF may be executed by larger GSDMD pores or eventually exacerbated membrane integrity and cell death (Figure 1, bottom panel).
The study by Dankers et al. touches upon additional facets of MIF secretion biology. Following up on their previous work showing that loss of autophagy further enhances LPS‐stimulated MIF secretion,21 they here confirm in their BMDM model that autophagy inhibition promotes MIF release similar to that of IL‐1β. Thus, autophagy inhibition may contribute to MIF release during pyroptosis, but it might also be one of the mechanisms relevant in constitutive MIF secretion under homeostatic conditions. However, the actual pore or channel that enables MIF secretion at baseline remains unknown (Figure 1, upper panel). Alternatively, a vesicular mechanism or exosome‐mediated export of MIF as hypothesized previously may be responsible for MIF release under such conditions, which would be in line with coordinated secretion/reuptake processes ensuring a homeostatic extracellular MIF pool, and the growth factor‐like characteristics of MIF that Dankers et al. also speculate about.
In summary, the current study fills a gap in our understanding about the release mechanisms of MIF proteins during various stages of inflammation. Most importantly, it adds necroptosis and pyroptosis to the list of relevant pathways mediating MIF release from macrophages, offers initial mechanistic explanations and suggests a staggered sequence of cell activation from homeostasis to marked inflammation and damage‐associated cell stress (Figure 1). Some interesting mechanistic questions remain to be clarified in the future. For example: (i) what is the role of p1159 in these release processes? (ii) what is the molecular identity of the pore? and (iii) what are the relative contributions of the different mechanisms in early stage, low‐grade inflammatory stimulation versus elevated inflammation versus exuberant hyperstimulation associated with cell damage? The answers will also help to address open questions regarding LPS‐triggered MIF secretion from fully viable macrophages such as RAW264.7 or peritoneal exudate macrophages versus BMDMs or human monocytes,6, 22 MIF release mechanisms in other nonimmune cell types or the intriguing observation from single‐cell secretion profiles, suggesting the existence of stimulus‐dependent MIF secretion‐competent cells.23 Regardless of these open mechanistic aspects, our gained knowledge of the MIF secretion mechanism furthers the concept that MIF is a key upstream mediator of monocyte/macrophage inflammation.
中文翻译:
再次探讨巨噬细胞迁移抑制因子的分泌机制,欢迎来到“ UPS俱乐部”。
细胞因子是调节细胞间通讯的蛋白质介质。它们协调免疫反应,并在多种疾病中发挥关键作用,例如炎性和自身免疫性疾病,心血管疾病和癌症。因此,了解它们的表达和分泌机制对于鉴定阻断病原性细胞因子释放的治疗策略是重要且有条件的。大多数细胞因子带有一个N端信号肽,并通过常规的内质网(ER)-高尔基体途径分泌。但是,许多重要的炎症介质,包括白介素(IL)-1细胞因子家族,均不表达信号肽,而是通过其他途径释放,这些途径统称为非常规蛋白分泌(UPS)。巨噬细胞迁移抑制因子(MIF)是发现的最古老的细胞因子之一。它是先天免疫的关键上游调节剂和重要的宿主防御蛋白,而失调的MIF是急性和慢性炎症,动脉粥样硬化,类风湿关节炎和系统性红斑狼疮(SLE)的关键介质。1-3 MIF是一种“非典型”细胞因子;这一概念不仅指其在各个王国中的高度进化保守性,而且还指诸如N端互变异构酶口袋,其趋化因子样行为和CXC趋化因子受体的参与,缺乏信号肽和丰富的胞质表达等分子特征。作为推测的其他细胞内功能。[4]实际上,以前曾有人建议通过非常规的蛋白质分泌途径释放MIF,[ 5]但缺少最终证据,确切的机制尚不完全清楚。Dankers等人的这项研究。在其分泌机制中增加了一个重要的新方面,将MIF释放与坏死病和核苷酸结合域,含有3个(NLRP3)炎性小体依赖性细胞凋亡的富含亮氨酸重复序列(NLR)家族吡喃域的过程联系起来。6这些数据有助于将MIF确立为“蛋白质的非常规蛋白质分泌UPS俱乐部”的成员。
MIF被发现是T细胞和巨噬细胞的细胞因子,但现在很清楚,它在大多数细胞类型中都广泛表达。MIF的大量胞质储存池和1-10 ng mL -1范围内的基线(非病理性)循环MIF水平被认为是其非典型细胞因子谱的一部分。在发炎的情况下,循环中的MIF水平迅速而急剧地增加,达到500-1500 ng mL -1的水平。疾病条件下释放的MIF蛋白的实际局部组织水平定义不清,但在体外基于细胞培养的研究表明,相似的升高率高达10到100倍。尽管MIF分泌研究主要是在免疫细胞中进行的,但很明显,分泌还可以响应内皮细胞和血小板,实质细胞(例如心肌细胞,血管平滑肌细胞和成纤维细胞)的多种刺激而发生细胞。MIF释放上调的潜在机制是非常规的,并且内质网/高尔基体独立性最初是由于MIF序列中缺少信号肽而引起的,并且通过使用经典ER /高尔基体途径抑制剂的研究实验性地证明了这一点。7、8蛋白质相互作用研究和药理抑制剂的应用牵涉到AIF转运蛋白ABCA1,信号小体亚基JAB1 / CSN5和胞质高尔基体相关蛋白p115与MIF的非常规蛋白分泌途径有关。9 JAB1 / CSN5被认为具有螯合功能,p115促进MIF分泌,但尚不清楚这些蛋白是直接还是间接参与,以及介导MIF从细胞内区室释放或转运到细胞外的实际机制尚不清楚空间仍然不确定。4研究永生化小鼠骨髓衍生的巨噬细胞(BMDM)和人THP-1单核细胞样细胞,Dankers等人。现在提供的证据表明,单核细胞/巨噬细胞释放的MIF可能是由坏死,坏死性坏死和发烧介导的。6
过氧化氢,乙醇或紫外线导致坏死,并在6小时的治疗时间内导致细胞外MIF升高3到100倍。坏死诱导的MIF释放在心肌细胞和嗜中性粒细胞10、11之前已经显示,实际上,对于非典型的细胞因子(如MIF),其半结构性表达并以相对较高的浓度储存在细胞内实际上是可以预期的。实际上,坏死刺激巨噬细胞后的释放模式反映了乳酸脱氢酶(LDH)作为细胞内持家蛋白的代表。6
坏死病是一种坏死的程序化形式,也被认为是调节性炎症细胞死亡。与坏死不同,坏死性坏死期间细胞膜的通透性受到严格调节,并由caspase依赖性受体相互作用激酶-1(RIPK1)/ RIPK3 /核糖体/混合谱系激酶结构域类假激酶(MLKL)介导的孔形成介导。丹克斯等。分别用脂多糖(LPS)/泛半胱天冬酶抑制剂Z-VAD和TNF抑制剂/凋亡抑制(IAP)蛋白BV-6 / Z-VAD家族治疗其骨髓源巨噬细胞和THP-1单核细胞,以诱导坏死病。他们观察到MIF迅速大量释放,这特别取决于坏死性信号传导机制,这一点已通过使用RIPK1抑制剂necrostatin-1进行的实验证实。这是第一个因果关系将MIF从免疫细胞释放与坏死联系起来的报道(图1)。有趣的是,以前已经提出了MIF与坏死性病之间的联系。我们先前显示,MIF信号传导会增加心脏成纤维细胞的坏死性病变,但在急性肾损伤的情况下,可防止肾小管上皮细胞坏死性病变。12,13巴恩斯等。肝细胞中乙醇诱导的受体相互作用激酶-3 RIPK3依赖性MIF释放涉及坏死性坏死。14结合这些研究,Dankers等人的最新工作。可能提示MIF在坏死性细胞死亡过程中具有更广泛的作用。坏死病迅速促进了MIF的释放,而细胞外MIF又控制了这种程序性炎症细胞死亡过程。
细胞凋亡是程序性坏死细胞死亡的另一种形式,与坏死细胞相似,但仅限于免疫细胞,具有更高的炎症潜能。关键的机制差异是:(i)NLRP3炎性体(“缩酶体”)通过细胞内危险信号而不是细胞外触发物引发;(ii)半胱天冬酶的参与(人类中的caspase-1 / 4/5和小鼠的caspase-11),导致IL-1家族细胞因子的加工和激活,以及(iii)胃泌素D(GSDMD)的蛋白水解生成,是坏死病中MLKLs混合谱系激酶结构域样假激酶与干细胞凋亡过程中的成孔蛋白。15 Dankers等。LPS引发,钾离子载体和NLRP3激活剂尼日利亚菌素的组合在其巨噬细胞模型中触发了NLRP3介导的凋亡。他们观察到了在这些条件下MIF的明显释放,并通过NLRP3特异性抑制剂MCC950验证了细胞凋亡的特异性,该抑制剂废除了MIF的释放。虽然MIF以前与NLRP3相关的炎症有关(图1),但Dankers等人的最新论文也是如此。在免疫学和细胞生物学是第一报告建立NLRP3炎性活性和分泌MIF之间机械因果关系。相同的作者先前发现,MIF充当NLRP3的上游激活剂,从而增强IL-1β的分泌。16出人意料的是,这是由细胞内MIF直接介导的,这是NLRP3和中间丝蛋白波形蛋白之间相互作用所必需的。康等人。在狼疮自身免疫的背景下,研究人员发现了MIF在人类单核细胞中上游控制NLRP3的另一种机制。他们表明,U1小核苷酸核糖核酸免疫复合物是MIF分泌的特定触发因素,随后以CD74依赖性方式激活NLRP3,caspase-1和下游IL-1β。17因此,NLRP3的这种上游控制机制是自分泌细胞外MIF的功能(图1)。此外,在以前的iTRAQ质谱分泌组筛选中,MIF与炎性体之间存在潜在的联系,这一现象已经变得显而易见,其中MIF是与caspase-1共分泌的最高级蛋白质之一。尽管没有采取后续的生物化学方法来证实MIF与caspase-1相互作用和/或MIF的分泌是因caspase-1依赖性的,但该研究表明NLRP3炎性体与MIF的分泌有关18(图1)。连同Dankers等人目前的工作,这些研究将MIF确立为NLRP3活性的上游调节剂和NLRP3依赖性焦磷酸化细胞因子分泌机制的新靶标。
这就引出了一个有趣的问题,丹克斯等人都没有。其他任何一项研究都尚未解决:从机械上讲,在尸体坏死或发烧过程中(或通过其他建议的途径;图1)释放的细胞内MIF蛋白如何穿过质膜屏障到达细胞外区室?可以在坏死病发作期间释放细胞内表达的与危险相关的分子模式(DAMPs,也称为alarmis)的膜通透孔是MLKL。焦磷酸细胞活化中发炎的巨噬细胞从发炎的巨噬细胞中释放IL-1β,其他IL-1细胞因子和DAMPs是通过GSDMD形成的膜孔介导的。根据最近的概念,“小” GSDMD孔可以充当通道,促进具有完整质膜的活细胞释放IL-1β和其他小的无前导细胞因子,19,20尚未发现在活化的单核细胞/巨噬细胞中执行MIF释放的孔分子。图1(中间面板)总结了机械选项。如Dankers等。如果发现MIF释放是NLRP3依赖性的,那么GSDMD孔可能会与MIF释放同时形成,并且未来的研究将需要确定GSDMD是否可以充当MIF传输孔。或者,先前牵涉的ABCA1转运蛋白或未知的通道/孔可能充当MIF释放孔。先前对药理抑制剂的研究也暗示了一种水泡出口机制,其细节仍不清楚。5此外,Dankers等人进行的一些实验。提示MIF释放可能在发烧和/或坏死病的更晚期“溶解”阶段显着增加。在这一阶段,MIF的膜移位可能是通过更大的GSDMD孔执行的,或者最终加剧了膜的完整性和细胞死亡(图1,底部)。
Dankers等人的研究。涉及MIF分泌生物学的其他方面。跟踪他们先前的工作表明自噬的丧失进一步增强了LPS刺激的MIF分泌,21他们在他们的BMDM模型中证实自噬抑制会促进MIF释放,类似于IL-1β。因此,自噬抑制作用可能会导致焦磷酸化过程中MIF的释放,但是它也可能是体内稳态条件下本构MIF分泌相关的机制之一。但是,使MIF在基线分泌的实际孔或通道仍然未知(图1,上图)。或者,如先前假设的水泡机制或外来体介导的MIF出口可能是这种情况下MIF释放的原因,这与协调的分泌/再摄取过程相一致,从而确保体内细胞外MIF池稳定,并具有生长因子样特征Dankers等人的MIF的内容。也推测一下。
总而言之,当前的研究填补了我们对炎症各个阶段中MIF蛋白释放机制的理解的空白。最重要的是,它在介导巨噬细胞释放MIF的相关途径列表中增加了坏死病和发烧现象,提供了初步的机理解释,并提出了从稳态到显着的炎症和损伤相关细胞应激的交错细胞激活序列(图1)。将来有一些有趣的机制问题需要澄清。例如:(i)p115 9在这些发布过程中的作用是什么?(ii)孔的分子身份是什么?(iii)早期低度炎症刺激与炎症增强与过度刺激与细胞损伤相关?这些问题的答案也将有助于关于从完全可行的巨噬细胞,如RAW264.7或腹腔渗出巨噬细胞LPS触发MIF分泌地址开放的问题与的BMDM或人单核细胞,6,22个在其他非免疫细胞类型的MIF释放机制或耐人寻味的观察单细胞分泌谱,表明存在刺激依赖性MIF分泌能力细胞。23无论这些开放的机制如何,我们对MIF分泌机制的了解进一步深化了MIF是单核细胞/巨噬细胞炎症的关键上游介质的概念。

























 京公网安备 11010802027423号
京公网安备 11010802027423号