当前位置:
X-MOL 学术
›
ChemPhysChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Hydrogen bond structure and low-frequency dynamics of electrolyte solutions: hydration numbers from ab initio water reorientation dynamics and dielectric relaxation spectroscopy.
ChemPhysChem ( IF 2.9 ) Pub Date : 2020-08-31 , DOI: 10.1002/cphc.202000498 Seonmyeong Kim 1, 2 , Xiangwen Wang 3 , Jeongmin Jang 1, 2 , Kihoon Eom 1, 2 , Simon L Clegg 4 , Gun-Sik Park 1, 2 , Devis Di Tommaso 3
ChemPhysChem ( IF 2.9 ) Pub Date : 2020-08-31 , DOI: 10.1002/cphc.202000498 Seonmyeong Kim 1, 2 , Xiangwen Wang 3 , Jeongmin Jang 1, 2 , Kihoon Eom 1, 2 , Simon L Clegg 4 , Gun-Sik Park 1, 2 , Devis Di Tommaso 3
Affiliation
|
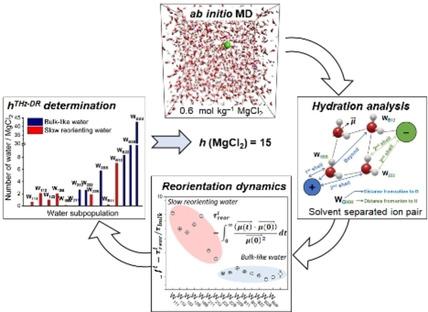
We present an atomistic simulation scheme for the determination of the hydration number (h) of aqueous electrolyte solutions based on the calculation of the water dipole reorientation dynamics. In this methodology, the time evolution of an aqueous electrolyte solution generated from ab initio molecular dynamics simulations is used to compute the reorientation time of different water subpopulations. The value of h is determined by considering whether the reorientation time of the water subpopulations is retarded with respect to bulk‐like behavior. The application of this computational protocol to magnesium chloride (MgCl2) solutions at different concentrations (0.6–2.8 mol kg−1) gives h values in excellent agreement with experimental hydration numbers obtained using GHz‐to‐THz dielectric relaxation spectroscopy. This methodology is attractive because it is based on a well‐defined criterion for the definition of hydration number and provides a link with the molecular‐level processes responsible for affecting bulk solution behavior. Analysis of the ab initio molecular dynamics trajectories using radial distribution functions, hydrogen bonding statistics, vibrational density of states, water‐water hydrogen bonding lifetimes, and water dipole reorientation reveals that MgCl2 has a considerable influence on the hydrogen bond network compared with bulk water. These effects have been assigned to the specific strong Mg‐water interaction rather than the Cl‐water interaction.
中文翻译:

氢键结构和电解质溶液的低频动力学:从头开始的水重新取向动力学和介电弛豫光谱学中的水合数。
我们基于水偶极子重取向动力学的计算,提出了一种原子模拟方案,用于确定电解质水溶液的水合数(h)。在这种方法中,从头算分子动力学模拟生成的电解质水溶液的时间演化用于计算不同水亚群的重新定向时间。h的值是通过考虑水亚群的重新定向时间是否相对于块状行为而受到延迟来确定的。将该计算方案应用于不同浓度(0.6–2.8 mol kg -1)的氯化镁(MgCl 2)溶液时,得到h这些值与使用GHz至THz介电弛豫光谱法获得的实验水合值非常吻合。该方法之所以具有吸引力,是因为它基于定义水合数的明确标准,并提供了与影响整体溶液行为的分子水平过程的联系。使用径向分布函数,氢键统计,状态振动密度,水-水氢键寿命和水偶极重取向分析从头算分子动力学轨迹,发现与大体积水相比,MgCl 2对氢键网络有相当大的影响。这些效应被分配给特定的强镁水相互作用,而不是氯水相互作用。
更新日期:2020-10-19
中文翻译:
氢键结构和电解质溶液的低频动力学:从头开始的水重新取向动力学和介电弛豫光谱学中的水合数。
我们基于水偶极子重取向动力学的计算,提出了一种原子模拟方案,用于确定电解质水溶液的水合数(h)。在这种方法中,从头算分子动力学模拟生成的电解质水溶液的时间演化用于计算不同水亚群的重新定向时间。h的值是通过考虑水亚群的重新定向时间是否相对于块状行为而受到延迟来确定的。将该计算方案应用于不同浓度(0.6–2.8 mol kg -1)的氯化镁(MgCl 2)溶液时,得到h这些值与使用GHz至THz介电弛豫光谱法获得的实验水合值非常吻合。该方法之所以具有吸引力,是因为它基于定义水合数的明确标准,并提供了与影响整体溶液行为的分子水平过程的联系。使用径向分布函数,氢键统计,状态振动密度,水-水氢键寿命和水偶极重取向分析从头算分子动力学轨迹,发现与大体积水相比,MgCl 2对氢键网络有相当大的影响。这些效应被分配给特定的强镁水相互作用,而不是氯水相互作用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号