Journal of Molecular Graphics and Modelling ( IF 2.9 ) Pub Date : 2020-08-29 , DOI: 10.1016/j.jmgm.2020.107726 Isil Ilgaz Aysan 1 , Taylan Gorkan 1 , Ilkay Ozdemir 1 , Yelda Kadioglu 1 , Gökhan Gökoğlu 2 , Ethem Aktürk 3
|
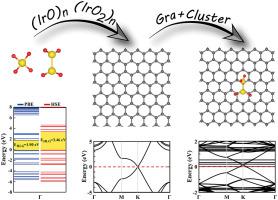
In this study, we investigated and revealed the electronic properties, geometric structures and binding behavior of small (IrO)n and (n = 1–5) clusters within first principles calculations based on the density functional theory. The electronic and magnetic properties of small nanoclusters displayed significant size dependency due to strong quantum confinement effect. Moreover we considered the binding and structural modification of the clusters on graphene surface as a substrate. The cohesive energy per atom of isolated clusters increased with size of the cluster n. This shows that the increase in coordination number results in a more stable nanocluster with increased number of saturated bonds. Pristine (IrO)n and clusters presented different structural motives at equilibrium. The ground states of (IrO)n and clusters considered in this study were all magnetic except for (IrO)4, , and . HOMO-LUMO gap EHLG values displayed large variations due to size of the cluster, hence bond saturation. The structural configurations of free standing nanoclusters are slightly modified, when adsorbed on graphene. The adsorption behavior of a cluster on graphene was improved by an applied electric field yielding larger binding energy and larger charger transfer. We observed that electronic and magnetic ground state of the clusters strongly depend on optimized structural configuration for both bare and adsorbed on graphene monolayer.
中文翻译:
石墨烯上吸附的氧化铱团簇的电子结构,内聚力和磁性。
在这项研究中,我们调查并揭示了小(IrO)n和铁的电子性质,几何结构和结合行为。(n = 1–5)在基于密度泛函理论的第一性原理计算中成簇。由于强大的量子约束效应,小纳米团簇的电子和磁性表现出明显的尺寸依赖性。此外,我们认为石墨烯表面上的团簇作为底物的结合和结构修饰。孤立簇的每个原子的内聚能随簇n的大小而增加。这表明配位数的增加导致具有增加的饱和键数目的更稳定的纳米簇。原始(IrO)n和集群在平衡时表现出不同的结构动机。(IrO)n和除(IrO)4外,本研究中考虑的团簇都是磁性的,和 。由于簇的大小,HOMO-LUMO间隙E HLG值显示出较大的变化,因此键饱和。当独立的纳米簇吸附在石墨烯上时,其结构构型会稍作改变。通过施加电场产生更大的结合能和更大的电荷转移,改善了簇在石墨烯上的吸附行为。我们观察到,簇的电子和磁性基态在很大程度上取决于裸石墨烯和吸附在石墨烯单层上的优化结构构型。
























 京公网安备 11010802027423号
京公网安备 11010802027423号