当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experiments and Direct Dynamics Simulations that Probe η2-Arene/Aryl-Hydride Equilibria of Tungsten Benzene Complexes
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2020-08-26 , DOI: 10.1021/jacs.0c08032 Jacob A Smith 1 , Anna Schouten 2 , Justin H Wilde 1 , Karl S Westendorff 1 , Diane A Dickie 1 , Daniel H Ess 2 , W Dean Harman 1
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2020-08-26 , DOI: 10.1021/jacs.0c08032 Jacob A Smith 1 , Anna Schouten 2 , Justin H Wilde 1 , Karl S Westendorff 1 , Diane A Dickie 1 , Daniel H Ess 2 , W Dean Harman 1
Affiliation
|
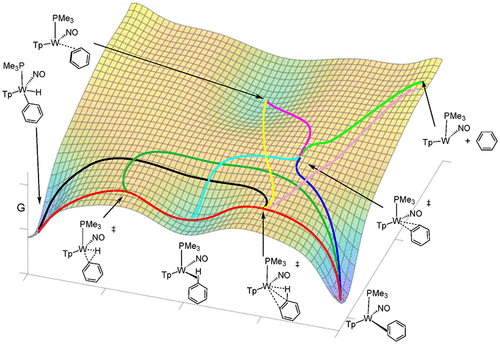
Key steps in the functionalization of unactivated arenes often involve its dihapto-coordination by a transition metal followed by insertion into the C-H bond. But rarely are the η2-arene and aryl hydride species in measurable equilibrium. In this study, the benzene/phenyl hydride equilibrium is explored for the {WTp(NO)(PBu3)} (Bu = n-butyl) system as a function of temperature, solvent, ancillary ligand, and arene substituent. Both face-flip and ring-walk isomerizations are identified through spin saturation exchange measurements, which both appear to operate through scission of a C-H bond. The effect of either an electron-donating or electron-withdrawing substituent is to increase the stability of both arene and aryl hydride isomers. Crystal structures, electrochemical measurements, and extensive NMR data, further support these findings. Static density functional theory (DFT) calculations of the benzene-to-phenyl hydride landscape suggest a single linear sequence for this transformation involving a sigma complex and oxidative cleavage transition state. Static DFT calculations also identified an η2-coordinated benzene complex in which the arene is held more loosely than in the ground state, primarily through dispersion forces. Although a single reaction pathway was identified by static calculations, quasiclassical direct dynamics simulations identified a network of several reaction pathways connecting the η2-benzene and phenyl hydride isomers, due to the relatively flat energy landscape.
中文翻译:

探索钨苯配合物的 η2-芳烃/芳基-氢化物平衡的实验和直接动力学模拟
未活化芳烃功能化的关键步骤通常涉及其通过过渡金属进行二联配位,然后插入 CH 键。但很少有 η2-芳烃和芳基氢化物处于可测量的平衡状态。在本研究中,探索了 {WTp(NO)(PBu3)}(Bu = 正丁基)系统的苯/苯氢化物平衡作为温度、溶剂、辅助配体和芳烃取代基的函数。面翻转和环走异构化都是通过自旋饱和交换测量来识别的,这两者似乎都是通过 CH 键的断裂来进行的。给电子或吸电子取代基的作用是增加芳烃和芳基氢化物异构体的稳定性。晶体结构、电化学测量和广泛的核磁共振数据进一步支持了这些发现。苯-苯氢化物景观的静态密度泛函理论 (DFT) 计算表明,这种转化有一个单一的线性序列,包括 sigma 复合物和氧化裂解过渡态。静态 DFT 计算还确定了一种 η2 配位苯配合物,其中芳烃比基态更松散,主要是通过色散力。尽管静态计算确定了单一反应途径,但由于能量格局相对平坦,准经典直接动力学模拟确定了连接 η2-苯和苯氢化异构体的几个反应途径网络。
更新日期:2020-08-26
中文翻译:
探索钨苯配合物的 η2-芳烃/芳基-氢化物平衡的实验和直接动力学模拟
未活化芳烃功能化的关键步骤通常涉及其通过过渡金属进行二联配位,然后插入 CH 键。但很少有 η2-芳烃和芳基氢化物处于可测量的平衡状态。在本研究中,探索了 {WTp(NO)(PBu3)}(Bu = 正丁基)系统的苯/苯氢化物平衡作为温度、溶剂、辅助配体和芳烃取代基的函数。面翻转和环走异构化都是通过自旋饱和交换测量来识别的,这两者似乎都是通过 CH 键的断裂来进行的。给电子或吸电子取代基的作用是增加芳烃和芳基氢化物异构体的稳定性。晶体结构、电化学测量和广泛的核磁共振数据进一步支持了这些发现。苯-苯氢化物景观的静态密度泛函理论 (DFT) 计算表明,这种转化有一个单一的线性序列,包括 sigma 复合物和氧化裂解过渡态。静态 DFT 计算还确定了一种 η2 配位苯配合物,其中芳烃比基态更松散,主要是通过色散力。尽管静态计算确定了单一反应途径,但由于能量格局相对平坦,准经典直接动力学模拟确定了连接 η2-苯和苯氢化异构体的几个反应途径网络。

























 京公网安备 11010802027423号
京公网安备 11010802027423号