当前位置:
X-MOL 学术
›
J. Phys. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Modulating excited‐state intramolecular proton transfer of 2‐(5‐(4‐carboxyphenyl)‐2‐hydroxyphenyl)benzothiazole depending on substituents: A DFT/TD‐DFT study
Journal of Physical Organic Chemistry ( IF 1.8 ) Pub Date : 2020-07-20 , DOI: 10.1002/poc.4109 Mei Ni 1 , Hua Fang 1
Journal of Physical Organic Chemistry ( IF 1.8 ) Pub Date : 2020-07-20 , DOI: 10.1002/poc.4109 Mei Ni 1 , Hua Fang 1
Affiliation
|
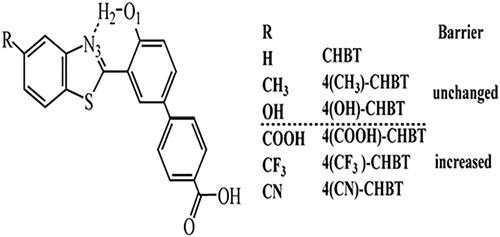
The OH‐based excited‐state intramolecular proton transfer (ESIPT) of 2‐(5‐(4‐carboxyphenyl)‐2‐hydroxyphenyl)benzothiazole (CHBT) derivatives (4R‐CHBT) that were replaced by electron‐donating groups (CH3, OH) and electron‐withdrawing groups (COOH, CF3, CN) have been studied by the time‐dependent density functional theory (TD‐DFT) method. We analyzed structural parameters, infrared (IR) vibrational spectrum, frontier molecular orbitals (FMOs), and electronic spectra, which can describe the trend of ESIPT process. The constructed potential energy surfaces showed that the ESIPT potential energy barriers of 4R‐CHBT are in the range of 0.25 to 0.62 kcal/mol, which are just a little bigger than that value of CHBT. All the chosen substituents, no matter electron‐donating or electron‐withdrawing groups, make ESIPT processes slightly more difficult.
中文翻译:

根据取代基调节2-(5-(4-羧基苯基)-2-羟基苯基)苯并噻唑的激发态分子内质子转移:DFT / TD-DFT研究
将O H-基于激发态分子内质子转移(ESIPT)的2-(5-(4-羧基苯基)-2-羟基苯基)由给电子基团被替换的苯并噻唑(CHBT)衍生物(4R-CHBT)( CH 3, OH)和吸电子基团( COOH, CF 3,CN)已通过时变密度泛函理论(TD-DFT)方法进行了研究。我们分析了结构参数,红外(IR)振动光谱,前沿分子轨道(FMO)和电子光谱,可以描述ESIPT过程的趋势。构造的势能面表明4R-CHBT的ESIPT势能垒在0.25至0.62 kcal / mol的范围内,仅略大于CHBT的值。所有选择的取代基,无论是供电子基团还是吸电子基团,都使ESIPT过程变得更加困难。
更新日期:2020-07-20
中文翻译:
根据取代基调节2-(5-(4-羧基苯基)-2-羟基苯基)苯并噻唑的激发态分子内质子转移:DFT / TD-DFT研究
将O H-基于激发态分子内质子转移(ESIPT)的2-(5-(4-羧基苯基)-2-羟基苯基)由给电子基团被替换的苯并噻唑(CHBT)衍生物(4R-CHBT)( CH 3, OH)和吸电子基团( COOH, CF 3,CN)已通过时变密度泛函理论(TD-DFT)方法进行了研究。我们分析了结构参数,红外(IR)振动光谱,前沿分子轨道(FMO)和电子光谱,可以描述ESIPT过程的趋势。构造的势能面表明4R-CHBT的ESIPT势能垒在0.25至0.62 kcal / mol的范围内,仅略大于CHBT的值。所有选择的取代基,无论是供电子基团还是吸电子基团,都使ESIPT过程变得更加困难。

























 京公网安备 11010802027423号
京公网安备 11010802027423号