当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Optimized Proteomics Workflow for the Detection of Small Proteins.
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2020-08-19 , DOI: 10.1021/acs.jproteome.0c00286 Jürgen Bartel 1 , Adithi R Varadarajan 2 , Thomas Sura 1 , Christian H Ahrens 2 , Sandra Maaß 1 , Dörte Becher 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2020-08-19 , DOI: 10.1021/acs.jproteome.0c00286 Jürgen Bartel 1 , Adithi R Varadarajan 2 , Thomas Sura 1 , Christian H Ahrens 2 , Sandra Maaß 1 , Dörte Becher 1
Affiliation
|
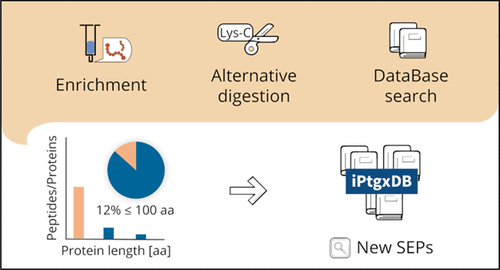
Small open reading frame encoded proteins (SEPs) gained increasing interest during the last few years because of their broad range of important functions in both prokaryotes and eukaryotes. In bacteria, signaling, virulence, and regulation of enzyme activities have been associated with SEPs. Nonetheless, the number of SEPs detected in large-scale proteome studies is often low as classical methods are biased toward the identification of larger proteins. Here, we present a workflow that allows enhanced identification of small proteins compared to traditional protocols. For this aim, the steps of small protein enrichment, proteolytic digest, and database search were reviewed and adjusted to the special requirement of SEPs. Enrichment by the use of small-pore-sized solid-phase material increased the number of identified SEPs by a factor of 2, and utilization of alternative proteases to trypsin reduced the spectral counts for larger proteins. The application of the optimized protocol allowed the detection of 210 already annotated proteins up to 100 amino acids (aa) length, including 16 proteins below 51 aa in the Gram-positive model organism Bacillus subtilis. Moreover, 12% of all identified proteins were up to 100 aa, which is a significantly larger fraction than that reported in studies involving traditional proteomics workflows. Finally, the application of an integrated proteogenomics search database and extensive subsequent validation resulted in the confident identification of three novel, not yet annotated, SEPs, which are 21, 26, and 42 aa long.
中文翻译:

优化的蛋白质组学工作流程,可检测小蛋白质。
在过去的几年中,小型开放阅读框编码的蛋白质(SEP)引起了人们越来越大的兴趣,因为它们在原核生物和真核生物中都有广泛的重要功能。在细菌中,信号传导,毒力和酶活性的调节与SEP有关。但是,由于传统方法倾向于识别较大的蛋白质,因此在大规模蛋白质组研究中检测到的SEP数量通常很少。在这里,我们提出了一种工作流程,与传统协议相比,该工作流程可以增强对小蛋白的鉴定。为此,对小蛋白质富集,蛋白水解消化和数据库搜索的步骤进行了审查,并根据SEP的特殊要求进行了调整。通过使用小孔径固相材料进行富集,将鉴定出的SEP数量增加了2倍 胰蛋白酶替代酶的使用和减少了较大蛋白质的光谱计数。通过优化方案的应用,可以检测到210种已注释的蛋白,最大长度为100个氨基酸(aa),包括革兰氏阳性模型生物体中51个氨基酸以下的16种蛋白枯草芽孢杆菌。此外,在所有鉴定出的蛋白质中,有12%的蛋白质高达100aa,这比涉及传统蛋白质组学工作流程的研究报告的分数高得多。最后,整合的蛋白质组学搜索数据库的应用和广泛的后续验证导致了对21个,26个和42个氨基酸长的3个新的,尚未注释的SEP的确定性鉴定。
更新日期:2020-10-02
中文翻译:
优化的蛋白质组学工作流程,可检测小蛋白质。
在过去的几年中,小型开放阅读框编码的蛋白质(SEP)引起了人们越来越大的兴趣,因为它们在原核生物和真核生物中都有广泛的重要功能。在细菌中,信号传导,毒力和酶活性的调节与SEP有关。但是,由于传统方法倾向于识别较大的蛋白质,因此在大规模蛋白质组研究中检测到的SEP数量通常很少。在这里,我们提出了一种工作流程,与传统协议相比,该工作流程可以增强对小蛋白的鉴定。为此,对小蛋白质富集,蛋白水解消化和数据库搜索的步骤进行了审查,并根据SEP的特殊要求进行了调整。通过使用小孔径固相材料进行富集,将鉴定出的SEP数量增加了2倍 胰蛋白酶替代酶的使用和减少了较大蛋白质的光谱计数。通过优化方案的应用,可以检测到210种已注释的蛋白,最大长度为100个氨基酸(aa),包括革兰氏阳性模型生物体中51个氨基酸以下的16种蛋白枯草芽孢杆菌。此外,在所有鉴定出的蛋白质中,有12%的蛋白质高达100aa,这比涉及传统蛋白质组学工作流程的研究报告的分数高得多。最后,整合的蛋白质组学搜索数据库的应用和广泛的后续验证导致了对21个,26个和42个氨基酸长的3个新的,尚未注释的SEP的确定性鉴定。

























 京公网安备 11010802027423号
京公网安备 11010802027423号