Chemical Physics ( IF 2.3 ) Pub Date : 2020-08-18 , DOI: 10.1016/j.chemphys.2020.110957 Yan Liu , Shu-Hua Xia , Yan Zhang
|
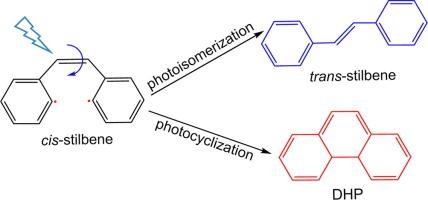
We combined the electronic structure calculations and OM2(orthogonalization model 2)/MRCI nonadiabatic surface-hopping dynamics simulations to scrutinize the photoisomerization mechanisms of cis-stilbene. The static electronic structure calculations showed that the irradiation of cis-stilbene populates the bright S1 state and we obtained three S1/S0 conical intersections (CIs) and proposed two nonadiabatic decay channels that efficiently de-excited to the ground state. In the first pathway, once excited to the S1 state, the cis-stilbene proceeds along S1 potential energy surface. Then, the molecule encounters S1S0-C CI, from which the system decays to S0 state. The other pathway involves the S1S0-D CI from where the S1 system hops to the ground state. In our simulations, we acquired cis, trans and DHP (4a,4b-dihydrophenanthrene) products and the ratio is estimated as 42:48:7. The present work show that the combination of electronic structure calculations and OM2/MRCI nonadiabatic dynamics can give detailed photochemical and photophysical information of stilbene systems.
中文翻译:
电子结构计算和非绝热表面跳跃动力学模拟的顺二苯乙烯分子的光化学和光物理性质
我们将电子结构计算与OM2(正交化模型2)/ MRCI非绝热表面跳跃动力学模拟相结合,研究了顺二苯乙烯的光异构化机理。静态电子结构计算表明,顺-二苯乙烯的辐照使明亮的S 1状态充满,我们获得了三个S 1 / S 0圆锥形交叉点(CIs),并提出了两个非绝热衰变通道,可以有效地将其激发为基态。在第一个途径中,一旦激发到S 1状态,顺式-二苯乙烯就沿着S 1前进势能面。然后,分子遇到S1S0-C CI,系统从中衰减到S 0状态。另一条路径涉及S1S0-D CI,S 1系统从此处跳至基态。在我们的模拟中,我们获得了顺式,反式和DHP(4a,4b-二氢菲)产物,该比例估计为42:48:7。目前的工作表明电子结构计算和OM2 / MRCI非绝热动力学的结合可以提供详细的光化学和光物理信息的系统。

























 京公网安备 11010802027423号
京公网安备 11010802027423号