当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Structure-Property Relationships and Machine Learning Models for Addressing CYP3A4-Mediated Victim Drug-Drug Interaction Risk in Drug Discovery.
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2020-08-14 , DOI: 10.1021/acs.molpharmaceut.0c00637 Bingjie Hu 1 , Xin Zhou 2 , Michael A Mohutsky 3 , Prashant V Desai 1
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2020-08-14 , DOI: 10.1021/acs.molpharmaceut.0c00637 Bingjie Hu 1 , Xin Zhou 2 , Michael A Mohutsky 3 , Prashant V Desai 1
Affiliation
|
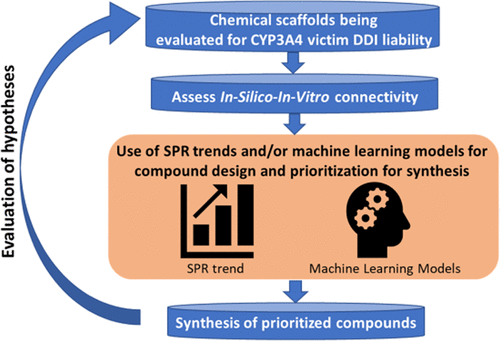
Among the FDA-approved small molecule drugs (2005–2016) that are primarily metabolized by cytochrome P450 (CYP), 64% are primarily metabolized by CYP3A4. As the proportion of an individual drug’s fraction metabolized through CYP3A4 increases, the risk for the drug to be a victim of an interaction with CYP3A4 inhibitors or inducers increases. Therefore, it is important to assess the extent of involvement of individual CYP enzymes in the overall clearance for a scaffold early in discovery and mitigate the CYP3A4-mediated victim–drug–drug interaction (DDI) risk, if warranted by the desired clinical profile of the drug. To mitigate the CYP3A4-mediated victim DDI risk in discovery, we analyzed the physicochemical properties of the CYP3A4 substrates and found that molecular weight was the property that provided the best separation of the CYP3A4 substrates from other CYP substrates. In addition, neutral and basic compounds with MW ≥ 360 g/mol tend to be primarily metabolized by CYP3A4, whereas acidic compounds with MW < 360 g/mol are most likely to be primarily metabolized by other CYP enzymes. We then developed Support Vector Machine based on fingerprints (SVM-FP) and Deep-Learning (DL) models to predict if a molecule will be primarily metabolized by CYP3A4. Our models were trained on 2306 compounds, which is the largest training set among published models for this endpoint. Both models showed positive predictive values (PPV) > 80% in predicting a CYP3A4 substrate on a prospective testing set. Given the high PPV of the models, project teams can confidently deprioritize compounds predicted to be CYP3A4 substrates to avoid the potential liability of CYP3A4 victim DDI. Teams can then focus time and resources on synthesizing compounds that are predicted to have a lower dependency on CYP3A4 metabolism and confirm that experimentally. Through such iterative in silico–in vitro learning circles, drug discovery teams can decide if metabolism through non-CYP3A4 pathways could be achieved in the SAR of a chemical series to mitigate the CYP3A4 victim DDI risk.
中文翻译:

解决药物发现中CYP3A4介导的受害人药物相互作用的结构-属性关系和机器学习模型。
在主要由细胞色素P450(CYP)代谢的FDA批准的小分子药物(2005-2016)中,有64%主要由CYP3A4代谢。随着通过CYP3A4代谢的单个药物级分比例的增加,该药物成为与CYP3A4抑制剂或诱导剂相互作用的受害者的风险增加。因此,重要的是评估早期发现的支架中单个CYP酶参与总体清除的程度,并减轻CYP3A4介导的受害者-药物-药物相互作用(DDI)的风险,如果所期望的临床特征所保证毒品。为减轻发现中CYP3A4介导的受害者DDI的风险,我们分析了CYP3A4底物的理化性质,发现分子量是提供CYP3A4底物与其他CYP底物最佳分离的性质。此外,MW≥360 g / mol的中性和碱性化合物倾向于主要通过CYP3A4代谢,而MW <360 g / mol的酸性化合物最有可能主要通过其他CYP酶代谢。然后,我们基于指纹(SVM-FP)和深度学习(DL)模型开发了支持向量机,以预测某个分子是否主要被CYP3A4代谢。我们的模型在2306种化合物上进行了训练,这是针对该终点的已发布模型中最大的训练集。两种模型在前瞻性测试组上预测CYP3A4底物时均显示阳性预测值(PPV)> 80%。鉴于模型的PPV高,项目团队可以放心地取消对预测为CYP3A4底物的化合物的优先级,从而避免CYP3A4受害者DDI的潜在责任。然后,研究小组可以将时间和资源集中在合成对CYP3A4代谢依赖性较低的化合物上,并通过实验进行确认。通过这样的迭代在计算机体外学习圈子中,药物发现小组可以决定是否可以通过化学系列SAR中的非CYP3A4途径实现代谢,以减轻CYP3A4受害者DDI的风险。
更新日期:2020-09-09
中文翻译:
解决药物发现中CYP3A4介导的受害人药物相互作用的结构-属性关系和机器学习模型。
在主要由细胞色素P450(CYP)代谢的FDA批准的小分子药物(2005-2016)中,有64%主要由CYP3A4代谢。随着通过CYP3A4代谢的单个药物级分比例的增加,该药物成为与CYP3A4抑制剂或诱导剂相互作用的受害者的风险增加。因此,重要的是评估早期发现的支架中单个CYP酶参与总体清除的程度,并减轻CYP3A4介导的受害者-药物-药物相互作用(DDI)的风险,如果所期望的临床特征所保证毒品。为减轻发现中CYP3A4介导的受害者DDI的风险,我们分析了CYP3A4底物的理化性质,发现分子量是提供CYP3A4底物与其他CYP底物最佳分离的性质。此外,MW≥360 g / mol的中性和碱性化合物倾向于主要通过CYP3A4代谢,而MW <360 g / mol的酸性化合物最有可能主要通过其他CYP酶代谢。然后,我们基于指纹(SVM-FP)和深度学习(DL)模型开发了支持向量机,以预测某个分子是否主要被CYP3A4代谢。我们的模型在2306种化合物上进行了训练,这是针对该终点的已发布模型中最大的训练集。两种模型在前瞻性测试组上预测CYP3A4底物时均显示阳性预测值(PPV)> 80%。鉴于模型的PPV高,项目团队可以放心地取消对预测为CYP3A4底物的化合物的优先级,从而避免CYP3A4受害者DDI的潜在责任。然后,研究小组可以将时间和资源集中在合成对CYP3A4代谢依赖性较低的化合物上,并通过实验进行确认。通过这样的迭代在计算机体外学习圈子中,药物发现小组可以决定是否可以通过化学系列SAR中的非CYP3A4途径实现代谢,以减轻CYP3A4受害者DDI的风险。


























 京公网安备 11010802027423号
京公网安备 11010802027423号