Analytical and Bioanalytical Chemistry ( IF 4.3 ) Pub Date : 2020-07-25 , DOI: 10.1007/s00216-020-02809-z Silvia Millán-Martín 1 , Craig Jakes 1, 2 , Sara Carillo 1 , Tom Buchanan 3 , Marc Guender 4 , Dan Bach Kristensen 5 , Trine Meiborg Sloth 5 , Martin Ørgaard 5 , Ken Cook 6 , Jonathan Bones 1, 2
|
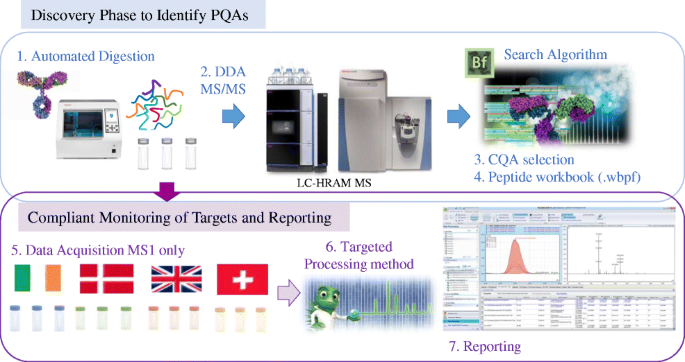
Peptide mapping analysis is a regulatory expectation to verify the primary structure of a recombinant product sequence and to monitor post-translational modifications (PTMs). Although proteolytic digestion has been used for decades, it remains a labour-intensive procedure that can be challenging to accurately reproduce. Here, we describe a fast and reproducible protocol for protease digestion that is automated using immobilised trypsin on magnetic beads, which has been incorporated into an optimised peptide mapping workflow to show method transferability across laboratories. The complete workflow has the potential for use within a multi-attribute method (MAM) approach in drug development, production and QC laboratories. The sample preparation workflow is simple, ideally suited to inexperienced operators and has been extensively studied to show global applicability and robustness for mAbs by performing sample digestion and LC-MS analysis at four independent sites in Europe. LC-MS/MS along with database searching was used to characterise the protein and determine relevant product quality attributes (PQAs) for further testing. A list of relevant critical quality attributes (CQAs) was then established by creating a peptide workbook containing the specific mass-to-charge (m/z) ratios of the modified and unmodified peptides of the selected CQAs, to be monitored in a subsequent test using LC-MS analysis. Data is provided that shows robust digestion efficiency and low levels of protocol induced PTMs.
中文翻译:
使用自动胰蛋白酶消化监测单克隆抗体产品质量属性的优化肽图工作流程的实验室间研究。
肽图分析是验证重组产物序列的一级结构和监测翻译后修饰 (PTM) 的监管预期。尽管蛋白水解消化已经使用了几十年,但它仍然是一个劳动密集型的过程,很难准确复制。在这里,我们描述了一种快速且可重复的蛋白酶消化方案,该方案使用磁珠上的固定化胰蛋白酶进行自动化,该方案已被纳入优化的肽图工作流程中,以显示跨实验室的方法可转移性。完整的工作流程有可能在药物开发、生产和 QC 实验室的多属性方法 (MAM) 方法中使用。样品制备工作流程很简单,非常适合没有经验的操作员,并通过在欧洲的四个独立地点进行样品消解和 LC-MS 分析进行了广泛的研究,以显示 mAb 的全球适用性和稳健性。LC-MS/MS 与数据库搜索一起用于表征蛋白质并确定相关产品质量属性 (PQA) 以进行进一步测试。然后通过创建包含所选 CQA 的修饰和未修饰肽的特定质荷 (m/z) 比的肽工作簿来建立相关关键质量属性 (CQA) 的列表,以便在后续测试中进行监控使用 LC-MS 分析。提供的数据显示强大的消化效率和低水平的协议诱导 PTM。


























 京公网安备 11010802027423号
京公网安备 11010802027423号