Current Computer-Aided Drug Design ( IF 1.7 ) Pub Date : 2021-06-30 , DOI: 10.2174/1573409916666200628104134 Rameez Jabeer Khan 1 , Rajat Kumar Jha 1 , Gizachew Muluneh Amera 1 , Jayaraman Muthukumaran 1 , Rashmi Prabha Singh 2 , Amit Kumar Singh 1
|
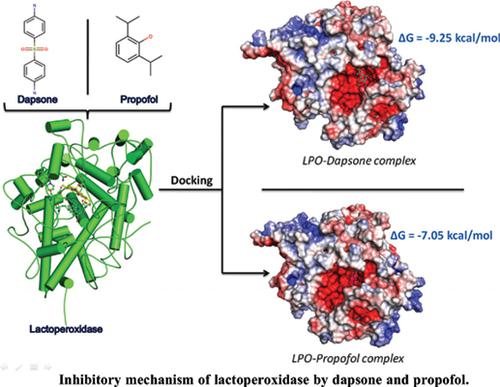
Introduction: Lactoperoxidase (LPO) is a member of the mammalian heme peroxidase family and is an enzyme of the innate immune system. It possesses a covalently linked heme prosthetic group (a derivative of protoporphyrin IX) in its active site. LPO catalyzes the oxidation of halides and pseudohalides in the presence of hydrogen peroxide (H2O2) and shows a broad range of the antimicrobial activity.
Methods: In this study, we have used two pharmaceutically important drug molecules, namely dapsone and propofol, which were earlier reported as potent inhibitors of LPO. At the same time, the stereochemistry and mode of binding of dapsone and propofol to LPO are still not known because of the lack of the crystal structures of LPO with these two drugs. In order to fill this gap, we utilized molecular docking and molecular dynamics (MD) simulation studies of LPO in its native and complex forms with dapsone and propofol.
Results: From the docking results, the estimated binding free energies (ΔG) of -9.25 kcal/mol (Ki = 0.16 μM) and -7.05 kcal/mol (Ki = 6.79 μM) were observed for dapsone, and propofol, respectively. The standard error of the Auto Dock program is 2.5 kcal/mol; therefore, molecular docking results alone were inconclusive.
Conclusion: To further validate the docking results, we performed MD simulation on unbound, and two drugs bounded LPO structures. Interestingly, MD simulations results explained that the structural stability of LPO-Propofol complex was higher than LPO-Dapsone complex. The results obtained from this study establish the mode of binding and interaction pattern of the dapsone and propofol to LPO as inhibitors.
中文翻译:
一项全面的计算机研究,旨在了解氨苯砜和丙泊酚对乳过氧化物酶的抑制机制
简介:乳过氧化物酶 (LPO) 是哺乳动物血红素过氧化物酶家族的成员,是一种先天免疫系统的酶。它在其活性部位具有共价连接的血红素辅基(原卟啉 IX 的衍生物)。LPO 在过氧化氢 (H 2 O 2 ) 存在下催化卤化物和拟卤化物的氧化,并显示出广泛的抗菌活性。
方法:在这项研究中,我们使用了两种药学上重要的药物分子,即氨苯砜和丙泊酚,它们早先被报道为有效的 LPO 抑制剂。同时,氨苯砜和丙泊酚与LPO的立体化学和结合方式仍不清楚,因为这两种药物缺乏LPO的晶体结构。为了填补这一空白,我们利用氨苯砜和丙泊酚对其天然和复杂形式的 LPO 进行分子对接和分子动力学 (MD) 模拟研究。
结果:根据对接结果,氨苯砜和丙泊酚的估计结合自由能 (ΔG) 分别为 -9.25 kcal/mol (Ki = 0.16 μM) 和 -7.05 kcal/mol (Ki = 6.79 μM)。Auto Dock 程序的标准误差为 2.5 kcal/mol;因此,单独的分子对接结果是不确定的。
结论:为了进一步验证对接结果,我们对未结合的和两种药物结合的 LPO 结构进行了 MD 模拟。有趣的是,MD模拟结果解释了LPO-丙泊酚复合物的结构稳定性高于LPO-氨苯砜复合物。从这项研究中获得的结果确立了氨苯砜和丙泊酚作为抑制剂与 LPO 的结合模式和相互作用模式。


























 京公网安备 11010802027423号
京公网安备 11010802027423号