当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
FT-IR and FT-Raman investigation, quantum chemical analysis and molecular docking studies of 5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2-yl)phenyl-4,5-dihydro-1H-pyrazol-1-yl]-1,3-thiazol-4(5H)-one
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.129070 K. Venil , A. Lakshmi , V. Balachandran , B. Narayana , Vinutha V. Salian
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2021-02-01 , DOI: 10.1016/j.molstruc.2020.129070 K. Venil , A. Lakshmi , V. Balachandran , B. Narayana , Vinutha V. Salian
|
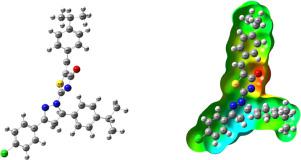
Abstract The FT-IR and FT-Raman spectra of 5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2-yl)phenyl-4,5-dihydro-1H-pyrazol-1-yl]-1,3-thiazol-4(5H)-one were recorded and analyzed. The fundamental vibrational wavenumbers, intensities of vibrational bands and the optimized geometrical parameters of the compound were evaluated using DFT (B3LYP) method with 6-31G, 6-31G(d,p) basis sets. Stability of the molecule arising from hyper conjugative interactions, charge delocalization has been analysed using natural bond orbital (NBO) analysis. Information about the size, shape, charge density distribution and site of chemical reactivity of the molecule has been obtained by mapping electron density iso-surface with molecular electrostatic potential (MEP) surface. The calculated HOMO-LUMO energies also show that the charge transfer occurs within the molecule. The global reactivity parameters which are obtained by frontier molecular orbital disclose that the molecule might be bioactive. To explain the chemical selectivity or the reactivity site in the molecule, the electron density-based local reactivity descriptors such as Fukui functions were calculated. The reduced density gradient of the title molecule was investigated by the interaction of the molecule. Molecular docking studies were also described. This study may also provide a further investigation of thiazole derivatives for pharmacological importance.
中文翻译:

5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2)的FT-IR和FT-Raman研究、量子化学分析和分子对接研究-yl)phenyl-4,5-dihydro-1H-pyrazol-1-yl]-1,3-thiazol-4(5H)-one
摘要 5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2-yl)phenyl-4的FT-IR和FT-Raman光谱,5-二氢-1H-吡唑-1-基]-1,3-噻唑-4(5H)-one 被记录和分析。使用DFT (B3LYP) 方法以6-31G、6-31G(d,p) 基组对化合物的基本振动波数、振动带强度和优化的几何参数进行评估。使用自然键轨道 (NBO) 分析法分析了由超共轭相互作用、电荷离域引起的分子的稳定性。通过用分子静电势 (MEP) 表面映射电子密度等值面,获得了有关分子大小、形状、电荷密度分布和化学反应位点的信息。计算的 HOMO-LUMO 能量还表明电荷转移发生在分子内。通过前沿分子轨道获得的全局反应性参数表明该分子可能具有生物活性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。
更新日期:2021-02-01
中文翻译:
5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2)的FT-IR和FT-Raman研究、量子化学分析和分子对接研究-yl)phenyl-4,5-dihydro-1H-pyrazol-1-yl]-1,3-thiazol-4(5H)-one
摘要 5-(4-Propan-2-yl)benzylidene)-2-[3-(4-chlorophenyl)-5[4-(propan-2-yl)phenyl-4的FT-IR和FT-Raman光谱,5-二氢-1H-吡唑-1-基]-1,3-噻唑-4(5H)-one 被记录和分析。使用DFT (B3LYP) 方法以6-31G、6-31G(d,p) 基组对化合物的基本振动波数、振动带强度和优化的几何参数进行评估。使用自然键轨道 (NBO) 分析法分析了由超共轭相互作用、电荷离域引起的分子的稳定性。通过用分子静电势 (MEP) 表面映射电子密度等值面,获得了有关分子大小、形状、电荷密度分布和化学反应位点的信息。计算的 HOMO-LUMO 能量还表明电荷转移发生在分子内。通过前沿分子轨道获得的全局反应性参数表明该分子可能具有生物活性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。为了解释分子中的化学选择性或反应性位点,计算了基于电子密度的局部反应性描述符,例如 Fukui 函数。通过分子的相互作用研究标题分子的密度梯度降低。还描述了分子对接研究。这项研究还可以进一步研究噻唑衍生物的药理学重要性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号