当前位置:
X-MOL 学术
›
Solid State Commun.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Electronic and optical properties of W-Sn-Z and W′-Sn-W′ monolayers using density functional theory
Solid State Communications ( IF 2.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.ssc.2020.114016 M. Barhoumi , N. Sfina , K. Lazaar , M. Said
Solid State Communications ( IF 2.1 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.ssc.2020.114016 M. Barhoumi , N. Sfina , K. Lazaar , M. Said
|
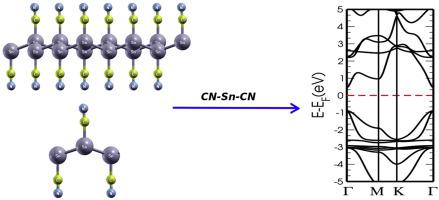
Abstract Similar to graphene, the subject of stanene has stolen significant recognition both in experimental and theoretical areas. Nevertheless, stanene is poor when put into practice because it has zero band-gap energy, therefore it is necessary to use a universal strategy to achieve a material with a large band-gap. Employing density functional theory, we study the structural, electronic, and optical properties such as dielectric function and absorption coefficient of stanene monolayer functionalized with chemical groups, i.e., W–Sn-Z, Cl–Sn–Br, and W′-Sn-W′ (where W = Cl, F, or OH; Z = CN; and W′ = CN, CH, or NH). Our calculations reveal that the band-gap energy at the high symmetry points tuned in a range of 1.019–1.751 eV, which is significantly higher than the stanene monolayer with hydrogenation (0.3 eV). Furthermore, we found that under an external electric field the electronic band structures of various compounds are changed down in energy producing in a semiconductor to metal transition. Furthermore, we have computed other properties like the absorption coefficient, the refractive index, and the conductivity of our systems.
中文翻译:

使用密度泛函理论 W-Sn-Z 和 W'-Sn-W' 单层的电子和光学性质
摘要 与石墨烯类似,stanene 主题在实验和理论领域都获得了重要的认可。然而,由于锡烯的带隙能量为零,因此在实际应用时效果不佳,因此需要采用通用策略来实现大带隙材料。利用密度泛函理论,我们研究了用化学基团(即 W-Sn-Z、Cl-Sn-Br 和 W'-Sn-)功能化的锡烯单层的结构、电子和光学性质,如介电函数和吸收系数。 W'(其中 W = Cl、F 或 OH;Z = CN;W' = CN、CH 或 NH)。我们的计算表明,高对称点的带隙能量在 1.019-1.751 eV 的范围内调整,这显着高于氢化的单层锡烯 (0.3 eV)。此外,我们发现在外部电场下,各种化合物的电子能带结构在半导体到金属的过渡中产生的能量发生变化。此外,我们还计算了其他属性,例如我们系统的吸收系数、折射率和电导率。
更新日期:2020-11-01
中文翻译:
使用密度泛函理论 W-Sn-Z 和 W'-Sn-W' 单层的电子和光学性质
摘要 与石墨烯类似,stanene 主题在实验和理论领域都获得了重要的认可。然而,由于锡烯的带隙能量为零,因此在实际应用时效果不佳,因此需要采用通用策略来实现大带隙材料。利用密度泛函理论,我们研究了用化学基团(即 W-Sn-Z、Cl-Sn-Br 和 W'-Sn-)功能化的锡烯单层的结构、电子和光学性质,如介电函数和吸收系数。 W'(其中 W = Cl、F 或 OH;Z = CN;W' = CN、CH 或 NH)。我们的计算表明,高对称点的带隙能量在 1.019-1.751 eV 的范围内调整,这显着高于氢化的单层锡烯 (0.3 eV)。此外,我们发现在外部电场下,各种化合物的电子能带结构在半导体到金属的过渡中产生的能量发生变化。此外,我们还计算了其他属性,例如我们系统的吸收系数、折射率和电导率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号