当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
σ-Hole interactions in small-molecule compounds containing divalent sulfur groups R1-S-R2.
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2020-08-07 , DOI: 10.1107/s2052520620008598 Albert S Lundemba 1 , Dikima D Bibelayi 1 , Peter A Wood 2 , Juliette Pradon 2 , Zéphyrin G Yav 1
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2020-08-07 , DOI: 10.1107/s2052520620008598 Albert S Lundemba 1 , Dikima D Bibelayi 1 , Peter A Wood 2 , Juliette Pradon 2 , Zéphyrin G Yav 1
Affiliation
|
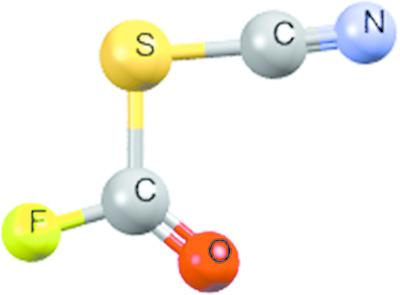
Hydrogen bonds, aromatic stacking contacts and σ‐hole interactions are all noncovalent interactions commonly observed in biological systems. Structural data derived from the Protein Data Bank showed that methionine residues can interact with oxygen atoms through directional S…O contacts in the protein core. In the present work, the Cambridge Structural Database (CSD) was used in conjunction with ab initio calculations to explore the σ‐hole interaction properties of small‐molecule compounds containing divalent sulfur. CSD surveys showed that 7095 structures contained R1—S—R2 groups that interact with electronegative atoms like N, O, S and Cl. Frequencies of occurrence and geometries of the interaction were dependent on the nature of R1 and R2, and the hybridization of carbon atoms in C,C—S, and C,S—S fragments. The most common interactions in terms of frequency of occurrence were C,C—S…O, C,C—S…N and C,C—S…S with predominance of Csp2. The strength of the chalcogen interaction increased when enhancing the electron‐withdrawing character of the substituents. The most positive electrostatic potentials (VS,max; illustrating positive σ‐holes) calculated on R1—S—R2 groups were located on the S atom, in the S—R1 and S—R2 extensions, and increased with electron‐withdrawing R1 and R2 substituents like the interaction strength did. As with geometric data derived from the CSD, interaction geometries calculated for some model systems and representative CSD compounds suggested that the interactions were directed in the extensions of S—R1 and S—R2 bonds. The values of complexation energies supported attractive interactions between σ‐hole bond donors and acceptors, enhanced by dispersion. The interactions of R1—S—R2 with large VS,max and nucleophiles with large negative VS,min coherently provided more negative energies. According to NBO analysis, chalcogen interactions consisted of charge transfer from a nucleophile lone pair to an S—R1 or S—R2 antibonding orbital. The directional σ‐hole interactions at R1—S—R2 can be useful in crystal engineering and the area of supramolecular biochemistry.
中文翻译:

含二价硫基团R1-S-R2的小分子化合物中的σ-孔相互作用。
氢键,芳香族堆积接触和σ-孔相互作用都是在生物系统中常见的非共价相互作用。来自蛋白质数据库的结构数据表明,蛋氨酸残基可以通过蛋白质核心中的定向S…O接触与氧原子相互作用。在当前工作中,剑桥结构数据库(CSD)与从头算计算结合使用,以探索含二价硫的小分子化合物的σ孔相互作用特性。CSD调查显示,7095个结构包含与电负性原子(如N,O,S和Cl)相互作用的R 1 -S- R 2基团。相互作用的发生频率和几何形状取决于分子的性质R 1和R 2以及C,CS和CS片段中碳原子的杂化。就发生频率而言,最常见的相互作用是C,C–S…O,C,C–S…N和C,C–S…S,优势为C sp 2。当增强取代基的吸电子特性时,硫族元素相互作用的强度增加。最正性静电电位(V S,最大计算;表示正σ孔)- [R 1 -S- - [R 2个基团分别位于上的S原子,在S- [R 1和S- [R 2名扩展,和具有增加的吸电子R 1和R 2取代基像相互作用强度一样。与从CSD导出的几何数据一样,为某些模型系统和代表性的CSD化合物计算的相互作用几何结构表明,相互作用的方向是S R 1和S R 2键的扩展。络合能的值支持了σ孔键供体与受体之间的吸引作用,并通过分散增强了相互作用。的相互作用- [R 1 -S- - [R 2与大V S,最大的亲核试剂与大的负和V S,分钟相干地提供了更多的负能量。根据NBO分析,硫族元素的相互作用包括从亲核孤对到S R 1或S R 2反键轨道的电荷转移。R 1 -S- R 2处的定向σ空穴相互作用可用于晶体工程和超分子生物化学领域。
更新日期:2020-08-07
中文翻译:
含二价硫基团R1-S-R2的小分子化合物中的σ-孔相互作用。
氢键,芳香族堆积接触和σ-孔相互作用都是在生物系统中常见的非共价相互作用。来自蛋白质数据库的结构数据表明,蛋氨酸残基可以通过蛋白质核心中的定向S…O接触与氧原子相互作用。在当前工作中,剑桥结构数据库(CSD)与从头算计算结合使用,以探索含二价硫的小分子化合物的σ孔相互作用特性。CSD调查显示,7095个结构包含与电负性原子(如N,O,S和Cl)相互作用的R 1 -S- R 2基团。相互作用的发生频率和几何形状取决于分子的性质R 1和R 2以及C,CS和CS片段中碳原子的杂化。就发生频率而言,最常见的相互作用是C,C–S…O,C,C–S…N和C,C–S…S,优势为C sp 2。当增强取代基的吸电子特性时,硫族元素相互作用的强度增加。最正性静电电位(V S,最大计算;表示正σ孔)- [R 1 -S- - [R 2个基团分别位于上的S原子,在S- [R 1和S- [R 2名扩展,和具有增加的吸电子R 1和R 2取代基像相互作用强度一样。与从CSD导出的几何数据一样,为某些模型系统和代表性的CSD化合物计算的相互作用几何结构表明,相互作用的方向是S R 1和S R 2键的扩展。络合能的值支持了σ孔键供体与受体之间的吸引作用,并通过分散增强了相互作用。的相互作用- [R 1 -S- - [R 2与大V S,最大的亲核试剂与大的负和V S,分钟相干地提供了更多的负能量。根据NBO分析,硫族元素的相互作用包括从亲核孤对到S R 1或S R 2反键轨道的电荷转移。R 1 -S- R 2处的定向σ空穴相互作用可用于晶体工程和超分子生物化学领域。


























 京公网安备 11010802027423号
京公网安备 11010802027423号