当前位置:
X-MOL 学术
›
Geochim. Cosmochim. Acta
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Transformation of calcite to fluorapatite at room temperature: Impact of initial phosphate and fluoride levels
Geochimica et Cosmochimica Acta ( IF 5 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.gca.2020.07.039 Aravinth Siva Subramaniam Ekamparam , Abhas Singh
Geochimica et Cosmochimica Acta ( IF 5 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.gca.2020.07.039 Aravinth Siva Subramaniam Ekamparam , Abhas Singh
|
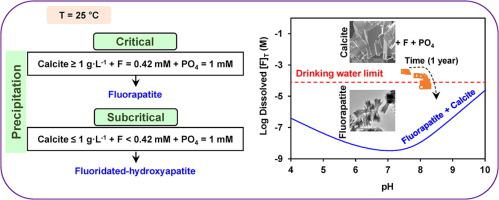
Abstract Geogenic fluoride (F) contamination in groundwater is a severe problem affecting millions of people across the world. For many F-contaminated aquifers, the solution saturation state is controlled by calcite and/or dolomite solubility. Recently, apatite-based ex situ F-immobilization techniques have gained attention as they minimize the release of secondary pollutants, otherwise attributed to conventional sorptive F-removal methods. Likewise, addition of phosphate (PO4) to F-contaminated subsurface environments could potentially facilitate immobilization of fluoride in sparingly soluble apatites. The success of these ex situ or in situ interventions would rely on careful understanding of the mechanisms behind F immobilization and the long-term stability of these immobilized forms. In this study, the impact of PO4 on the extent and forms of F uptake in the presence of calcite under conditions supersaturated with respect to fluorapatite (Ca5(PO4)3F; FA) were investigated. A year-long series of batch equilibration experiments with varying degrees of initial supersaturation, involving combinations of F (0.1–0.42 mM), PO4 (0.7–1 mM), and calcite (0.1–4 g·L−1), were conducted at room temperature. Total dissolved concentrations of F, Ca, and PO4 were analyzed with time. Changes in solution saturation state were evaluated within a thermodynamic modeling framework comprising an updated database of relevant aqueous and mineral solubility reactions. Additionally, surface complexation modeling was used to interpret F uptake on calcite. Results from the solution chemistry indicated that the system responded to relative kinetics of dissolution of calcite and precipitation of FA or fluoridated HA. System pH and Ca concentrations in the presence of phosphate were controlled by rapid calcite dissolution, which facilitated F uptake by creating conditions highly supersaturated with respect to FA. Formation of FA was suggested by the observed PO4: F molar uptake ratio (∼3), consistent with FA stoichiometry, and was confirmed with scanning electron microscopy-associated energy dispersive X-ray spectroscopy, X-ray diffraction, Fourier-transformed infrared spectroscopy, transmission electron microscopy-associated selected area diffraction, and X-ray photoelectron spectroscopy on solids collected at the end of the equilibration studies. Furthermore, stoichiometric PO4:F molar uptake was found only for systems containing concentrations of initial F, PO4, and calcite above certain critical levels (0.42 mM, 1 mM, and 1 g·L−1, respectively). Surface complexation modeling predictions suggested minimal adsorption of F and PO4 onto calcite for all conditions, irrespective of the initial concentrations. For sub-critical initial concentrations, F uptake probably occurred due to formation of fluoridated-hydroxyapatite (non-stoichiometric FA). Significantly, these immobilized F forms controlled the observed F and P concentrations below the respective drinking water and discharge limits for a year.
中文翻译:

室温下方解石向氟磷灰石的转化:初始磷酸盐和氟化物含量的影响
摘要 地下水中的地源氟化物 (F) 污染是一个严重的问题,影响着全世界数百万人。对于许多受 F 污染的含水层,溶液饱和状态由方解石和/或白云石溶解度控制。最近,基于磷灰石的非原位 F 固定技术受到关注,因为它们最大限度地减少了二次污染物的释放,否则归因于传统的吸附式 F 去除方法。同样,向 F 污染的地下环境添加磷酸盐 (PO4) 可能会促进氟化物在微溶磷灰石中的固定。这些非原位或原位干预的成功取决于对 F 固定背后的机制和这些固定形式的长期稳定性的仔细理解。在这项研究中,研究了在相对于氟磷灰石(Ca5(PO4)3F;FA)过饱和的条件下,在方解石存在下,PO4 对 F 吸收程度和形式的影响。进行了一系列具有不同初始过饱和度的为期一年的批次平衡实验,包括 F (0.1–0.42 mM)、PO4 (0.7–1 mM) 和方解石 (0.1–4 g·L-1) 的组合在室温下。随时间分析 F、Ca 和 PO4 的总溶解浓度。在热力学建模框架内评估溶液饱和状态的变化,该框架包括相关水和矿物溶解度反应的更新数据库。此外,表面络合模型用于解释方解石上的 F 吸收。溶液化学的结果表明,该系统响应方解石溶解和 FA 或氟化 HA 沉淀的相对动力学。磷酸盐存在下的系统 pH 值和 Ca 浓度由快速方解石溶解控制,这通过创造相对于 FA 高度过饱和的条件促进了 F 的吸收。观察到的 PO4:F 摩尔摄取比 (~3) 表明 FA 的形成,与 FA 化学计量一致,并通过扫描电子显微镜相关的能量色散 X 射线光谱、X 射线衍射、傅里叶变换红外光谱证实、透射电子显微镜相关的选区衍射和平衡研究结束时收集的固体的 X 射线光电子能谱。此外,化学计量的 PO4:仅在初始 F、PO4 和方解石浓度高于某些临界水平(分别为 0.42 mM、1 mM 和 1 g·L-1)的系统中发现 F 摩尔吸收。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。
更新日期:2020-11-01
中文翻译:
室温下方解石向氟磷灰石的转化:初始磷酸盐和氟化物含量的影响
摘要 地下水中的地源氟化物 (F) 污染是一个严重的问题,影响着全世界数百万人。对于许多受 F 污染的含水层,溶液饱和状态由方解石和/或白云石溶解度控制。最近,基于磷灰石的非原位 F 固定技术受到关注,因为它们最大限度地减少了二次污染物的释放,否则归因于传统的吸附式 F 去除方法。同样,向 F 污染的地下环境添加磷酸盐 (PO4) 可能会促进氟化物在微溶磷灰石中的固定。这些非原位或原位干预的成功取决于对 F 固定背后的机制和这些固定形式的长期稳定性的仔细理解。在这项研究中,研究了在相对于氟磷灰石(Ca5(PO4)3F;FA)过饱和的条件下,在方解石存在下,PO4 对 F 吸收程度和形式的影响。进行了一系列具有不同初始过饱和度的为期一年的批次平衡实验,包括 F (0.1–0.42 mM)、PO4 (0.7–1 mM) 和方解石 (0.1–4 g·L-1) 的组合在室温下。随时间分析 F、Ca 和 PO4 的总溶解浓度。在热力学建模框架内评估溶液饱和状态的变化,该框架包括相关水和矿物溶解度反应的更新数据库。此外,表面络合模型用于解释方解石上的 F 吸收。溶液化学的结果表明,该系统响应方解石溶解和 FA 或氟化 HA 沉淀的相对动力学。磷酸盐存在下的系统 pH 值和 Ca 浓度由快速方解石溶解控制,这通过创造相对于 FA 高度过饱和的条件促进了 F 的吸收。观察到的 PO4:F 摩尔摄取比 (~3) 表明 FA 的形成,与 FA 化学计量一致,并通过扫描电子显微镜相关的能量色散 X 射线光谱、X 射线衍射、傅里叶变换红外光谱证实、透射电子显微镜相关的选区衍射和平衡研究结束时收集的固体的 X 射线光电子能谱。此外,化学计量的 PO4:仅在初始 F、PO4 和方解石浓度高于某些临界水平(分别为 0.42 mM、1 mM 和 1 g·L-1)的系统中发现 F 摩尔吸收。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。表面络合模型预测表明,无论初始浓度如何,在所有条件下,F 和 PO4 在方解石上的吸附量都最小。对于亚临界初始浓度,F 吸收可能是由于氟化羟基磷灰石(非化学计量 FA)的形成而发生的。值得注意的是,这些固定化的 F 形式将观察到的 F 和 P 浓度控制在各自的饮用水和排放限值以下一年。

























 京公网安备 11010802027423号
京公网安备 11010802027423号